
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
ГЛАВА 1. ОБЗОР ЛИТЕРАТУРЫ
Бронхиальная астма (БА) является глобальной проблемой здравоохранения — по данным Всемирной организации здравоохранения (ВОЗ), в мире живет около 300 миллионов больных БА.
В настоящее время астма является наиболее распространенным хроническим заболеванием в промышленно развитых странах, встречающимся как у детей, так и у взрослых [81, 82]. Распространенность астмы резко возросла за последние 30 лет и достигает в некоторых регионах 30% среди взрослого населения. Неблагоприятная динамика распространенности БА связана с рядом факторов: ухудшение экологической ситуации, повышение частоты генетических дефектов, которые ассоциируются с развитием БА и ее тяжелым течением; распространение табакокурения и употребления алкоголя населением, профессиональные вредные факторы, нерациональное употребление лекарственных средств, самолечение, плохое питание и т. д. [58].
Немаловажным является факт экономического груза, возлагаемого на общество: стоимость болезни оценивается в 5,5 млрд. долларов в год [194].
По результатам популяционных исследований, в США число больных аллергией превышает 40 млн. человек. В структуре первичной заболеваемости по обращаемости заболевания органов дыхания стоят на первом месте [44]. По данным ВОЗ, более 20% людей на земном шаре страдает различными атопическими заболеваниями, одним из самых распространенных является аллергическая бронхиальная астма [78].
По мнению исследователей программы ISAAC (International Study of Asthma and Allergy in Childhood), различная частота симптомов заболевания в отдельных регионах связана с внешнесредовыми, социально-экономическими и этническими отличиями. Для большинства западных стран последние десятилетия ознаменовались изменением привычек и характера питания, нерациональным использованием антибиотиков, широким распространением гормональной контрацепции, увеличением частоты стрессов в обществе и количества курящих и, наконец, глобальным изменением климата с увеличением содержания различных токсинов в окружающей среде [52].
Важным эндогенным компонентом патогенеза при атопических заболеваниях является аномалия конституции (атопия) — генетическая склонность организма к гиперпродукции lgЕ в ответ на контакт с широко распространенными экзоаллергенами [9, 38, 73]. Однако известен и тот факт, что атопия, как иммунобиологический феномен, широко распространена в человеческой популяции.
В соответствии с последней номенклатурой аллергических болезней,
астму, опосредованную иммунологическими механизмами, следует называть аллергической астмой. Если доказано участие IgЕ антител в формировании астмы, то следует обозначать ее IgE-опосредованной аллергической астмой. В нашей стране эту форму бронхиальной астмы принято называть атопической [11]. Термин «атопия» следует использовать только после документированного подтверждения IgE сенсибилизации: выявлением специфических IgE антител в сыворотке крови или положительными результатами кожных тестов с аллергенами.
Бронхиальная астма — это хроническое воспалительное заболевание дыхательных путей, в котором принимают участие многие клетки и клеточные элементы. Хроническое воспаление обусловливает развитие бронхиальной гиперреактивности, которая приводит к повторяющимся эпизодам свистящих хрипов, одышки, чувства заложенности в груди и кашля, особенно по ночам или ранним утром. Эти эпизоды обычно связаны с распространенной, но изменяющейся по своей выраженности обструкцией дыхательных путей в легких, которая часто бывает обратимой либо спонтанно, либо под действием лечения [131].
Согласно современным представлениям, морфологической основой БА является хроническое воспаление бронхиальной стенки с повышением количества активированных эозинофилов, тучных клеток, Т-лимфоцитов в слизистой оболочке бронхов, утолщением базальной мембраны и последующим развитием субэпителиального фиброза [150].
В настоящее время признано, что все формы астмы связаны с воспалением в дыхательных путях, которое сохраняется даже при полном контроле БА [68].
Хроническое воспаление приводит к структурным изменениям в дыхательных путях, таких как фиброз, увеличение толщины гладкомышечного слоя дыхательных путей, гиперплазия секретирующих клеток и новых сосудов (ангиогенез), что может приводить к фиксированному сужению дыхательных путей [77].
Формирование хронического персистирующего воспаления в дыхательных путях опосредовано различными клетками и медиаторами, в том числе цитокинами и факторами роста [185].
В последние годы общепринятой считается теория, согласно которой аллергические заболевания обусловлены нарушениями регуляции в иммунной системе, связанной с активацией аллерген-специфических клонов Т-лимфоцитов хелперов, называемых Т-хелперами 2-го типа (Th2) [150].
Первичные сигналы, которые активируют Th2-клетки, неизвестны [83]. Возможно, эту роль первичного контакта между иммунной системой и внешними аллергенами играют дендритные клетки (ДК) и молекулы на поверхности антигенпрезентирующих клеток, в частности взаимодействие B72/CD28 [175]. Некоторые авторы отводят эту роль, переключение изотипов на Th2, интерлейкину-4 [229].
Активированная субпопуляция Т-хелперов 2 типа продуцирует ряд цитокинов: ИЛ-4, ИЛ-5, ИЛ-6, ИЛ-5 и ГМ-СФ (гранулоцитарный макрофагальный стимулирующий фактор); стимулирует пролиферацию и дифференцировку В-лимфоцитов, переключают синтез иммуноглобулинов в В-лимфоцитах на IgE и IgG4 [60, 82].
Ключевая роль именно CD4+ Th2-лимфоцитов в развитии аллергии продемонстрирована в опытах, где несенсибилизированным животным на фоне введения вызывающего астму антигена производили адаптивный перенос Th2-лимфоцитов, дифференцированных in vitro. В отличие от контрольных мышей, при введении Th2-клеток в легкие животных развивалась эозинофилия, увеличивалась секреция и нарастали симптомы гиперреактивности бронхов. Для того чтобы обеспечить развитие указанных симптомов аллергического воспаления, вначале при первичной встрече с антигеном должна произойти дифференцировка Th именно в направлении Th2 [150].
Избыток Th2 цитокинов, ИЛ-4 и ИЛ-13, по отношению к Th1-клеткам, продуцирующих ИНФ-γ, способствует доминированию механизмов аллергического воспаления при астме [151].
Активация субпопуляции Th2-лимфоцитов и продукция указанных цитокинов приводят к индукции и синтезу В-лимфоцитами IgE и IgG4, а также к активации и дифференциации тучных клеток и эозинофилов [20].
Образовавшиеся IgE и IgG4 фиксируются на поверхности клеток-мишеней аллергии I (тучных клетках и базофилах) и II порядка (эозинофилах, нейтрофилах, макрофагах, тромбоцитах) с помощью клеточных Fc‑рецепторов (FcЕRI). Основное количество тучных клеток и базофилов находится в подслизистом слое. При стимуляции аллергеном количество их возрастает в 10 раз [178].
Повторное взаимодействие IgE с антигеном приводит к перекрестному связыванию IgE с тучными клетками посредством рецепторов высокого сродства — FcЕRI, высвобождению вазоконстрикторных медиаторов,
синтезу простагландинов и лейкотриенов, а также к транскрипции цитокинов [150].
Наряду с активацией Тh2-клеток снижается функция субпопуляции Т‑лимфоцитов-хелперов 1 типа (Тh1). Thl-лимфоциты секретируют гамма-интерферон, который тормозит синтез реагинов (IgE) В-лимфоцитами [20].
В настоящее время многочисленными исследованиями показано, что ИЛ-4 является основным цитокином, участвующим в патогенезе атопической БА [148]. Именно ИЛ-4 содействует дифференцировке Th2-клеток и синтезу IgE [162]. Продукция ИЛ-4 может быть активирована стимуляцией рецепторов антигеном на Т-лимфоцитах и высокоаффинных Fсε рецепторов IgE, находящихся на тучных клетках и базофилах [194]. Интересно, что кортикостероиды повышают способность индуцировать синтез ИЛ-4 из CD4+ Т-клеток [84]. Кроме того, ИЛ-4 стимулирует экспрессию В-клетками молекулы главного комплекса гистосовместимости (HLA) II класса, В7-2, CD40, поверхностного IgM с низким сродством к рецептору, что приводит к повышению антигенпрезентирующей возможности В-клеток [108].
В ряде работ показано, что ИЛ-4 стимулирует бокаловидные клетки, вызывая гиперсекрецию слизи, и фибробласты, тем самым участвуя в ремоделировании дыхательных путей [114]. Другой потенциально важной ролью ИЛ-4 при АБА является его способность индуцировать экспрессию сосудистой молекулы клеточной адгезии-1 (VCAM-1) на эндотелиальных клетках, что приводит к адгезии Т-клеток, эозинофилов, базофилов и моноцитов на эндотелии [134].
С ИЛ-4 посредством ИЛ-4Ra рецепторов [106] тесно связан ИЛ-13. Показано, что уровни ИЛ-13 и ИЛ-4 повышаются после воздействия аллергена при АБА [174, 193].
В дыхательных путях больных астмой ИЛ-4 имеет решающее значение для дифференцировки наивных Th0-лимфоцитов в Th2-лимфоциты при первичной сенсибилизации, тогда как секреция ИЛ-13 более выражена во время вторичного воздействия антигена [160].
Еще одним важным цитокином в патогенезе АБА является ИЛ-5. Основной источник ИЛ-5 — активированные Тh2-лимфоциты, а также эозинофилы, тучные клетки, CD4+ и CD8+ Т-клетки [171]. ИЛ-5 имеет решающее значение в регуляции воспаления, индуцируя активацию эозинофилов и пролиферацию их предшественников [157]. Показано, что ИЛ-5 увеличивает гиперчувствительность дыхательных путей при АБА [236]. Кроме того, ИЛ-5 и ИЛ-9 способствуют пролиферации и дифференцировке предшественников эозинофилов в костном мозге [251].
В исследованиях по использованию анти-ИЛ-5 моноклональных антител (меполизумаб) у больных БА показана их чрезвычайная эффективность в снижении эозинофилии как в периферической крови, так и дыхательных путях [116].
Накопление эозинофилов в дыхательных путях является отличительной чертой АБА. Активированные эозинофилы выпускают широкий спектр цитотоксических белков и трансформированных факторов роста, которые вызывают повреждение эпителия, утолщение базальной мембраны эпителиальных и стромальных клеток, фиброз, ангиогенез и железистую гиперплазию [201]. Кроме того, они способствуют развитию спазма бронхов, выраженного воспалительного процесса в них, нарушению микроциркуляции, гиперсекреции слизи, развитию гиперреактивности бронхов. Эозинофилы сами по себе могут генерировать цитокины, такие как ИЛ-3, ИЛ-5 и ГМ-СФ [133].
Большую роль в развитии ранней и поздней астматической реакции также играют альвеолярные и бронхиальные макрофаги. В результате контакта аллергенов и Fc-рецепторов макрофагов они активируются, что приводит к продукции медиаторов: фактора, активирующего тромбоциты, лейкотриенов В4 (в небольших количествах С4 и Д4), 5-НЕТЕ (5-гидроксиэйкозо-тетраеновой кислоты — продукта липоксигеназного окисления арахидоновой кислоты), лизосомальных ферментов, нейтральных протеаз, бетаглюкуронидазы, PgD 2 [66].
Тучные клетки при контакте с антигеном высвобождают содержимое своих гранул: гистамин, лейкотриены (ЛТС4, ЛТД4, ЛТЕ4), простагландин Д, различные протеолитические ферменты. Кроме этих медиаторов, из тучных клеток выделяются также ИЛ-3, ИЛ-4, ИЛ-5, ИЛ-6, ИЛ-7, ИЛ-8, нейтрофильный и эозинофильный хемотаксический факторы, тромбоцитоактивирующий фактор, гранулоцитарно-макрофагальный колониестимулирующий фактор и фактор некроза опухолей [55].
В дополнение к активации тучных клеток через FcЕRI, IgE взаимодействует с другими рецепторами, в том числе CD23 (FcЕRII). CD23-IgE взаимодействие обеспечивает важный механизм, посредством которого аллерген-специфические IgE могут усиливать клеточный и гуморальный иммунный ответ при повторных встречах с аллергеном [81].
Дегрануляция тучных клеток и базофилов, активация эозинофилов с выделением большого количества медиаторов аллергии и воспаления приводят к развитию бронхоспазма, выраженной экспираторной одышки [201].
Работа и функция Th2-клеток находится под влиянием особых регуляторных Т-клеток — Тreg, которые обеспечивают иммунологическую толерантность, в том числе в респираторном тракте [201].
Выделяют натуральные и индуцибельные, или адаптивные, Тreg-клетки. Натуральные регуляторные (NТreg) клетки с фенотипом CD4+CD25+Foxр3+ составляют 1-5% периферических CD4+ Т-клеток. Они обеспечивают супрессию аутоиммунных Т-лимфоцитов и предупреждают образование Т‑эффекторных лимфоцитов [167].
Антиген-специфические адаптивные (индуцибельные) iТreg образуются в периферических тканях в результате иммунизации аллергеном или длительного нахождения аллергена в окружающей среде. Эти клетки способны вырабатывать ИЛ-10 и трансформирующий ростовой фактор-бета (ТРФ-β), которые ингибируют регуляторную функцию Th2 [168].
При АБА оба типа регуляторных пула клеток (NTreg и iTreg), введенных внутривенно, вызывали обратное развитие заболевания. Эффект iTreg зависел от высокого уровня ТРФ-β, ИЛ-10, ИНФ-γ. NTreg дифференцировались в iTreg (экспрессирующие ИЛ-10) в легких [65, 187].
Toll-подобные рецепторы (TLR) имеются на лейкоцитах, включая Т‑ и В-лимфоциты, клетках эпителия и эндотелия, фибробластах, мышечных и нервных клетках. Активация подобных рецепторов приводит к продукции цитокинов, развитию воспаления и аллергии [120, 243].
CD8+ T-лимфоциты вносят важный вклад в хронизацию процесса: ФНО-α и ИЛ-6, ИНФ-γ и ГМ-СФ увеличивают антигенпредставляющую способность макрофагов и экспрессию HLA-DR [163].
Несмотря на то что Th2-лимфоциты рассматриваются в качестве центральных клеток, запускающих аллергические воспалительные явления, нет ответа на вопрос, почему у больных АБА наблюдается увеличение частоты и тяжести обострений во время вирусных инфекций дыхательных путей (респираторно-синцитиальный вирус, риновирус), когда доминирующим звеном, как известно, являются Th1-лимфоциты [177].
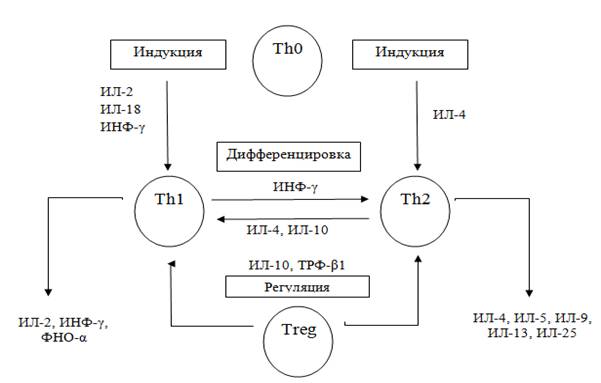
Рисунок 1 — Схема соотношения Th1/Th2 цитокинов [97, 126 с модификациями]
В ряде исследований предприняты попытки изучить также роль Th1-лимфоцитов и синтезируемых ими цитокинов в иммунопатогенезе аллергии. На экспериментальных моделях у животных и у больных БА в лаважной жидкости и в легочной ткани кроме увеличения Th2-цитокинов обнаружено увеличение содержания ИНФ-γ. Кроме того, отмечено, что концентрация ИНФ-γ повышается параллельно увеличению тяжести заболевания и коррелирует с количеством нейтрофилов [69]. Показано, что ИНФ-γ ингибирует эозинофилию, индуцированную аллергенами [161]. У больных астмой продукция как ИЛ-4, ИЛ-5, так и ИНФ-γ лимфоцитами лаважной жидкости и уровни ИНФ-γ в сыворотке периферической крови оказались повышены и коррелировали с тяжестью клинических проявлений [229, 230].
Приведенные результаты исследований взаимосвязи аллергии и уровней продукции ИНФ-γ свидетельствуют о возможном значении повышенного синтеза цитокинов Th1 для развития аллергических заболеваний. Кроме того, имеется ряд данных о возможном участии ИНФ-γ в поддержании аллергического воспаления в тканях, когда он действует совместно с цитокинами Th2- ответа, такими как ИЛ-4, ИЛ-5 и ИЛ-13 [146, 215].
Таким образом, цитокины Th1, в частности ИНФ-γ, могут усилить проявления аллергического воспаления в разных тканях, особенно на фоне уже сформировавшегося процесса. Видимо, регуляторные нарушения в иммунной системе, приводящие к развитию аллергии, могут быть объяснены сдвигом баланса Т-хелперных клонов в сторону активации Th2 и повышенного синтеза соответствующих цитокинов. Однако дальнейшее поддержание аллергического воспаления и формирование клинических симптомов может зависеть от цитокинов и Th2- и Th1-ответа.
Существует все больше доказательств, что астма — заболевание, связанное с окислительным стрессом [145]. Маркер окислительного стресса — 8-изопростан — специфичен для перекисного окисления липидов [196]. Недавние исследования показали более высокие уровни 8-изопростана в плазме крови, конденсате выдыхаемого воздуха и индуцированной мокроте больных астмой [159]. В нескольких исследованиях, проведенных на пациентах в обострении астмы, были зарегистрированы изменения в окислительном стрессе, в том числе в повышенном плазматическом уровне реактивных соединений тиобарбитуровой кислоты [156].
С-реактивный белок (СРБ), острофазный белок, является высокочувствительным системным маркером воспаления, инфекции и повреждения тканей [141, 213]. Рядом авторов установлена позитивная связь между астмой и повышением уровня СРБ в плазме крови [45, 76, 142]. Fujita с соавт., 2007, обнаружили, что сывороточные уровни СРБ могут быть связаны с обострениями астмы [106]. В соответствии с предыдущими отчетами, Judith C.W. Mak с соавторами, 2013, [182] описали значительно более высокие уровни СРБ в плазме на момент обострения заболевания и сохранение повышенных уровней в период ремиссии по сравнению со здоровыми, что подтверждает наличие хронического системного воспаления при БА [14].
Таким образом, механизмы, ведущие к усилению Th2-ответа, до сих пор остаются спорными. Th2-доминирование иммунных реакций может быть результатом супрессии Т-регуляторных клеток, а также Th1-лимфоцитов. Понимание иммунных механизмов, ответственных за атопическую БА, баланса Th1/Th2, продолжает быть актуальным направлением.
Изучение иммунных нарушений при АБА остается актуальным, так как контроль астмы — это не только достижение отсутствия клинических проявлений, улучшение показателей ФВД, но и контроль воспалительного процесса. Поэтому важен поиск биомаркеров, позволяющих определить тактику ведения и лечения больных АБА.
Кроме того, изучение цитокинов и/или их дисбаланса позволит выделить определенные фенотипы больных АБА для прогнозирования течения, степени тяжести заболевания, уровня контроля, выраженности иммунной недостаточности, а также подбора индивидуальной терапии.
Эндотелий — внутренняя выстилка сосудов — состоит приблизительно из 1-6·1013 клеток. Эндотелий интимы сосудов выполняет барьерную, секреторную, гемостатическую, вазотоническую функции, играет важную роль в процессах воспаления и ремоделирования сосудистой стенки.
Дисфункция эндотелия имеет значение в развитии ремоделирования сосудов, внутрисосудистой активации тромбоцитов и лейкоцитов [19].
Эндотелиальную дисфункцию (ЭД) можно определить как неадекватное (увеличенное или сниженное) образование в эндотелии различных биологически активных веществ. К факторам риска повреждения эндотелия относятся гиперхолестеринемия, гипергомоцистеинемия (ГГЦ), повышенный уровень цитокинов (ИЛ-1β, ФНО-α, ИЛ-8) [3, 26].
Эндотелий участвует в регуляции системного и легочного сосудистого тонуса посредством образования и высвобождения вазодилататоров и вазоконстрикторов, в частности эндотелина-1 и эндотелий-зависимого расслабляющего фактора — оксида азота (NO) [7, 225, 244]. Установлено, что эндотелий сосудов не просто образует барьер между кровью и гладкомышечными клетками сосудов, но и обеспечивает динамическое равновесие вазодилатирующих и вазоконстрикторных факторов, регулирует процессы гемостаза, влияет на сосудистую проницаемость и участвует в иммунном ответе организма [99, 245]. Кроме того, клетки эндотелия находятся в динамическом взаимодействии с гормональными и клеточными медиаторами, берущими начало в кровотоке и сосудистой стенке [4, 128]. Эндотелий вырабатывает вазодилататоры и антиагреганты (оксид азота (NO), брадикинин, простациклин, простагландин Е2, эндотелиальный фактор гиперполяризации), вазоконстрикторы и проагреганты (эндотелин-1 (ЭТ-1), ангиотензин ІІ (АТ ІІ), серотонин, простагландин F2α, лейкотриены С4, Д4, тромбоксан А2), гепарин, активаторы плазминогена, факторы роста [24, 25, 64, 195]. Функции NO включают обеспечение низкого сосудистого тонуса в покое, ингибирование агрегации и адгезии тромбоцитов, адгезии лейкоцитов и моноцитов, миграции и пролиферации гладкомышечных клеток [217].
Дисфункция эндотелия резко меняет направление его эндокринной активности на противоположную: образуются вазоконстрикторы, эндотелины, коагулянты, нарушается соотношение между NO (антиагрегантом, антикоагулянтом, вазодилятатором) и пероксинитратом-метаболитом NO, увеличивающим уровень окислительного стресса, что приводит к различным патофизиологическим реакциям [7].
Точные механизмы развития эндотелиальной дисфункции при БА недостаточно изучены, ключевое значение имеет ишемия/гипоксия тканей и сосудистой стенки, свободнорадикальное повреждение, действие цитокинов, гипергомоцистеинемия, эндогенные и экзогенные интоксикации,
возрастные изменения. Одной из причин нарушения регионарного кровообращения и микроциркуляции является дисфункция эндотелия, которая может приводить к спазму сосудов, усиленным тромбообразованию и адгезии лейкоцитов к эндотелию [61, 166].
Первый этап «каскада адгезии» — обратимое «привязывание» и «катание» лейкоцитов на эндотелии, опосредованное веществами группы селектина эндотелиальных клеток, такими как Е-селектин, Р-селектин, L‑селектин, и их лигандами на лейкоцитах.
Второй этап — стойкая адгезия лейкоцитов на эндотелии, осуществляется при участии молекул адгезии ICAM-1 (межклеточная молекула адгезии) и VCAM-1 (сосудисто-клеточная молекула адгезии) на эндотелиальных клетках и их соответствующими лигандами — антигенами LFA-1 (лейкоцитарный функциосвязанный антиген-1) и VLA-4 (очень поздний активированный антиген-4) на лейкоцитах [70].
В процессе адгезии и последующей миграции в субэндотелиальный слой лейкоциты активизируются и высвобождают протеазы и свободные
радикалы, приводящие к деградации (гидролизу) нормальной архитектоники сосудистой стенки. Развивается асептическое воспаление стенки сосуда, которое в условиях БА приобретает хронический характер. Это приводит к изменению эндотелий-зависимой вазодилатации, что является ключевым моментом в формировании ЭД [241].
Визуализация сосудов в ранней фазе ЭД часто демонстрирует неповреж-
денную сосудистую стенку, подтверждая тот факт, что макроскопические изменения сосудов видны не сразу [249].
В последнее десятилетие исследователи подчеркивают повреждающее действие провоспалительных цитокинов на эндотелий сосудов, запускающих каскад процессов от локальной вазоконстрикции и высвобождения факторов роста до процессов ремоделирования сосудистой стенки [26]. Доказано, что дисфункция эндотелия является обязательным компонентом практически всех сердечно‑сосудистых заболеваний. Однако в последние годы появились данные, свидетельствующие об участии эндотелия легочных сосудов в формировании легочной гипертензии [221].
В связи с этим особый интерес представляет вопрос о соотношении иммуновоспалительной активации и состояния сосудистого эндотелия у больных БА.
Клетки, инфильтрирующие слизистую бронхов при БА, способны также производить активные формы кислорода [37]. Известно, что повышение уровня частиц активного кислорода (ROS), особенно супероксид-аниона, вызывает нарушение синтеза и активности NО. В результате NO теряет такие свойства, как блокирование экспрессии адгезивных молекул эндотелия — ICAM-1, VCAM-1, PECAM-1, E-селектина и хемотаксических пептидов моноцитов, уменьшение агрегации, прилипания и инфильтрации сосудистой стенки нейтрофилами и моноцитами, торможение агрегации и адгезии тромбоцитов [180]. Еще один механизм снижения синтеза NO — разобщение эндотелиальной NO-синтазы (eNOS) на фоне окислительного стресса и продуцирование супероксид-аниона вместо NO [235]. Второй аспект
действия ROS — индукция экспрессии ICAM-1, VCAM-1, PECAM-1, что приводит к адгезии моноцитов, лимфоцитов на эндотелиальной стенке [181]. Более того, избыточное образование супероксидных и гидроксильных радикалов инициирует окисление липопротеидов низкой плотности (ЛПНП). Перекисно-модифицированные ЛПНП в силу своей токсичности повреждают эндотелиальный покров артерий [93] и приобретают способность секретировать биологически активные соединения (факторы роста, хемотоксины, митогены). Эти соединения стимулируют миграцию из медии в интиму гладкомышечных клеток и фибробластов, их пролиферацию и синтез соединительной ткани. Также ROS регулируют экспрессию различных факторов роста и ростовых протоонкогенов (c-Myc, c-Fos и c-Jun), что приводит к пролиферации и миграции в интиму артерий гладкомышечных клеток и усилению продукции ими коллагена и эластина [206]. Все вышеперечисленные механизмы в конечном итоге и приводят к развитию эндотелиальной дисфункции.
Морфологически у пациентов с БА отмечается повышение сечения подслизистого слоя сосудов, увеличение числа сосудов в стенках дыхательных путей, утолщение интимы [143, 203, 250]. Подобные элементы ремоделирования выявляются уже в детском возрасте при легком течении БА [108].
Считается, что после повреждения тканей лейкоциты выходят за предел сосудистого русла и инфильтрируют окружающие ткани. В процессе их миграции ключевую роль играет формирование контакта между лейкоцитами и эндотелиальными клетками, который обеспечивается молекулами клеточной адгезии (cell adgesion molecules — CAMs).
Процесс адгезии связан с появлением на клетках эндотелия молекул адгезии (Е-селектина и VCAM-1), а на эозинофилах и других клетках воспаления — соответствующих рецепторов для адгезивных молекул [127]. Экспрессия молекул адгезии на эндотелии усиливается действием цитокинов — ФНО-α и ИЛ-4, которые продуцируются тучными клетками. Так, ИЛ-4 усиливает экспрессию молекул адгезии, VCAM-1 в эндотелиальных и эпителиальных клетках дыхательных путей. В то же время ИЛ-1 и ФНО-α увеличивают экспрессию ICAM-1 как в сосудистом эндотелии, так и в эпителии дыхательных путей [239].
Связь между воспалением дыхательных путей и изменениями в капиллярах при БА была описана ранее [245].
Многие посредники и факторы роста, которые имеют прямое отношение к воспалению дыхательных путей при астме, например, гистамин, простагландины, лейкотриены и цитокины, могут одновременно вызывать ангиогенез и повышение проницаемости сосудов [158]. Однако роль многих из этих факторов часто нельзя трактовать однозначно, поскольку она основана на исследованиях, проведенных на животных или in vitro.
Поврежденные эндотелиальные клетки высвобождают медиаторы воспаления и различные молекулы адгезии, такие как межклеточные и сосудистые молекулы клеточной адгезии (ICAM-1, VCAM-1, PECAM-1), P‑ и E-селектины, необходимые для закрепления лейкоцитов к стенке сосуда, и известные как маркеры эндотелиальной дисфункции при воспалительных состояниях [94, 170].
VCAM-1 — vascular cellular adhesion molecule-1 — молекула адгезии сосудистого эндотелия 1 типа, экспрессируется на эндотелиальных клетках при стимуляции воспалительными цитокинами [176]. Функцией VCAM-1 является обеспечение адгезии лимфоцитов, моноцитов и эозинофилов к активированному эндотелию через α4β1 и α4β7 интегрины с последующей их экстравазацией и миграцией в очаг воспаления [239].
Е-селектин (ELAM-1, LECAM-2) экспрессируется также исключительно на эндотелии, обеспечивает адгезию лейкоцитов (нейтрофилов, моноцитов и субпопуляции Т-клеток) к активированному сосудистому эндотелию в начальных фазах воспаления [188, 234].
Особенностью именно этих молекул является их индуцибельная экспрессия, то есть только при активации эндотелиальных клеток провоспалительными цитокинами. Экспрессия VCAM-1 достигает максимума через 6-12 часов и длительно держится на высоком уровне [125]. Максимальная экспрессия Е-селектина достигается быстрее — через 4-6 часов, и менее пролонгирована (резко снижается после достижения пика).
Увеличенные уровни этих молекул могут свидетельствовать о повреждении эндотелия воспалительным процессом. Показано, что активация лейкоцитарно-эндотелиального взаимодействия сохраняется и вне обострения БА при различной степени тяжести заболевания [240].
Высокие уровни растворимого Е-селектина отражают его повышенную экспрессию, поэтому эта молекула может быть использована в качестве маркера персистирующего воспаления. В исследовании была обнаружена корреляционная взаимосвязь между высокими уровнями Е-селектина и тяжестью течения БА [117].
В экспериментальных условиях доказано, что при аллергической астме sЕ-селектин и sVCAM экспрессируются только эндотелием в отсутствии других коморбидных состояний [117]. При изучении sЕ-селектина и sVCAM у больных с атопической БА выявлено повышение этих молекул в период обострения заболевания [125], тогда как данные о концентрации их в межприступный период отсутствуют.
Известно, что молекулы VCAM-1 и ICAM-1 тесно связаны по структуре и функции. Тем не менее VCAM-1 уникален тем, что его экспрессия ограничивается в основном областью поражения, тогда как ICAM-1 экспрессируется в переходной области и непораженных участках эндотелия [212]. Эта разница, возможно, свидетельствует о различной функции в инициации поражения. В своем исследовании Myron I. с соавторами, изучая роль растворимых форм ICAM-1 и VCAM-1 на доклиническом уровне атеросклероза, показал доминирующую роль VCAM-1 в инициации атеросклероза [63].
Не менее важной и интересной является роль sCD31/sPECAM-1 как
маркера эндотелиальной дисфункции. Роль РЕСАМ-1 — трансмембранная передача сигнала межклеточного взаимодействия, что приводит к тромбозу, диапедезу лейкоцитов через сосудистую стенку в очаг воспаления [27]. Как показано в ранее проведенных исследованиях, в качестве одного из диагностических тестов, характеризующих состояние эндотелия сосудистой стенки, можно рассматривать изменение уровня sCD31 в плазме крови [41]. Полученные результаты выявили достоверное повышение sCD31/sPECAM-1 в плазме крови у больных БА в сравнении с контрольной группой, и наиболее высокие значения sCD31 регистрировались у пациентов с легким течением заболевания [27].
В отечественной литературе для изучения эндотелиальной дисфункции чаще используются стабильные метаболиты оксида озота, эндотелин-1,
десквамированные эндотелиоциты, активированный фактор Виллебранда [22, 103].
Методика определения состояния эндотелия с помощью ультразвука высокого разрешения была предложена в 1992 г. Celermajer D.S. [204]. Ее основой является измерение реакции плечевой артерии на реактивную гиперемию. Метод механической стимуляции эндотелия периферических артерий усиленным кровотоком после артериальной окклюзии дает представление о состоянии эндотелийзависимой вазодилатации (ЭЗВД) [204]. В последние годы рядом авторов установлено нарушение этого состояния у взрослых пациентов с хронической бронхолегочной патологией [121]. B. Wolff et al., 2007, показали, что ЭЗВД плечевой артерии отражает реактивность сосудов малого круга кровообращения [153].

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 |
