

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Нейрональное повреждение при постреанимационной болезни носит многофакторный характер и развивается в момент остановки кровообращения, в процессе проводимой сердечно-легочно-мозговой реанимации, а также в периоде восстановления самостоятельного кровообращения:
- период ишемии – аноксии в момент отсутствия кровообращения во время клинической смерти (no-flow); период гипоперфузии – гипоксии при искусственном поддержании кровообращения в процессе сердечно-легочно-мозговой реанимации (low-flow), поскольку максимально возможный уровень сердечного выброса достигает только 25 % от исходного; период реперфузии, состоящий из последовательно развивающихся фаз: no-reflow, следующей затем фазы гиперемии и последующей глобальной и мультифокальной гипоперфузии.
При этом подавляющее большинство процессов нейронального повреждения происходят не в момент остановки кровообращения или СЛР, а при реперфузии.
Клинически постреанимационное повреждение головного мозга проявляется различной степенью выраженности неврологической дисфункцией вплоть до комы и смерти мозга, а также персистирующим вегетативным состоянием. Если через 72 часа, после остановки кровообращения, неврологический дефицит составляет меньше 5 баллов по шкале ком Глазго, в отсутствие двигательной реакции на болевое раздражение или зрачкового рефлекса в отсутствие гипотермии, введения седативных средств и релаксантов, то это является предиктором развития персистирующего вегетативного состояния. Своевременное выявление поддающихся лечению неврологических расстройств, например, судорог, имеет большое значение. Распознание судорог может представлять трудности, особенно при назначении миорелаксантов, поэтому важным инструментом диагностики для таких пациентов является электроэнцефалограмма.
Выделяют три типа восстановления неврологического статуса в постаноксическом периоде:
Восстановление происходит после непродолжительного (3 часа) периода отсутствия сознания и характеризуется быстрой нормализацией адекватной психической деятельности в течение 24 часов после клинической смерти – у большинства (70%) пациентов. После выхода из острого патологического состояния у 50% больных развивается неврозоподобный синдром, кратковременные судороги, нейроциркуляторная дистония, рассеянная мелкоочаговая симптоматика. Задержанное восстановление функций ЦНС. Нарушение сознания (сомноленция, сопор, кома различной степени) может продолжаться в течение многих суток и зависит от развития отека головного мозга. У этих пациентов развиваются выраженные неврологические проявления в отдаленном периоде (2-3 месяца). Причем наиболее частым проявлением является неврозоподобный синдром в виде астении и раздражительной слабости. Из психических нарушений наиболее часто встречаются интеллектуально-мнестические расстройства (15,6 %).Патофизиология постаноксической энцефалопатии
В периоде острой аноксии существенное нарушение биоэнергетики клеток головного мозга является пусковым механизмом всех патологических процессов периода аноксии.
На фоне постаноксических вторичных нарушений перфузии мозга активация комплекса факторов в остром и подостром периодах приводит к диффузно-очаговым некротическим и апоптозным повреждениям части нейронов мозга, повышение активации компенсаторно-восстановительных процессов – к существенной реорганизации межнейронных взаимоотношений сохранившихся нейронов в отдаленном периоде.
Нормальное функционирование нейронов обеспечивается постоянной высокой активностью множества АТФаз, сохраняющих ионный гомеостаз, необходимый для функционирования нейронов и формирования их потенциала действия, в связи с чем более 70 % АТФ расходуется на поддержание транспортных трансмембранных систем. Дисфункция этих систем приводит к нарушению ионного гомеостаза и последующему запуску трех основных механизмов аноксического повреждения клеток головного мозга: свободнорадикального, кальцийзависимого и фосфолипазного. Эти механизмы по мере своей реализации в конечном итоге приводят к гибели клеток мозга путем некроза или апоптоза.
Так, компенсаторная активация гликолиза ведет к накоплению до токсического уровня лактата и вторичной блокаде многих ферментативных систем за счет увеличения внутриклеточной концентрации Н+, понижения НАДН, НАДФН в ткани мозга. Увеличение концентрации стимулирует процессы превращения АМФ в аденозин - инозин - гипоксантин, который активирует ксантиноксидазу и способствует образованию супероксидных радикалов.
В постреанимационном периоде гипергликемия приводит к более длительному неполному восстановлению рН ткани мозга и выраженному невосстановлению АТФ и креатинфосфата. Применение инсулина при гипергликемии позволяет значительно улучшить динамику восстановления рН, креатинфосфата, АТФ.
Нарушения биоэнергетики и ионного гомеостаза нейронов центральной нервной системы в период аноксии приводят к значительным изменениям их нейромедиаторного обмена, что в дальнейшем определяет судьбу нейронов при возобновлении их функционирования. Одним из таких механизмов является токсическое повреждение нейронов избыточным количеством возбуждающих нейромедиаторов. Именно формирование генераторов патологически усиленного возбуждения является ключевым звеном в процессе образования устойчивых патологических систем мозга и может лежать в основе отдаленной постаноксической энцефалопатии.
Выделяют две группы патологических механизмов, последовательно включающихся в результате увеличения внеклеточной концентрации возбуждающих нейромедиаторов.
Первая группа механизмов связана с ранним острым набуханием и отеком ткани мозга, они реализуются в момент аноксической деполяризации, являются обратимыми и неселективными, характерны для всех нейронов.
Механизмы второй группы включаются в результате повышения концентрации свободных ионов Са2+ в цитозоле нейронов в момент аноксической деполяризации, реализуются в течение длительного периода, являются необратимыми и селективными, приводя к гибели нейронов в течение 1–7 суток после аноксии. Таким образом, на фоне отека головного мозга включаются Са2+-зависимые патологические механизмы, которые перестраивают режим функционирования нейронов в постаноксическом периоде. У части селективно чувствительных нейронов это завершается их гибелью. Данная селективная чувствительность нейронов объясняется постаноксическим увеличением передачи возбуждающих импульсов через их синапсы, что сопровождается снижением порога возбудимости и гиперактивностью нейронов на фоне их неадекватного биоэнергетического обеспечения.
Развитие ССВО, пусковым механизмом которого является ишемия и реперфузия, дополнительно обусловливает повреждающее действие (рис. 2).
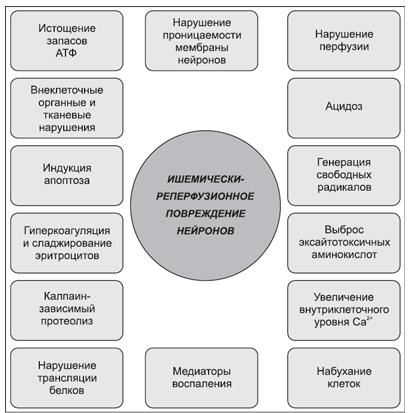
Рисунок 2. Механизмы ишемически-реперфузионного повреждения головного мозга при постреанимационной болезни (по A. E. Muniz, 2005)
В постреанимационном периоде выделяют следующие стадии нарушения перфузии головного мозга после восстановления самостоятельного кровообращения:
1. Начальное развитие мультифокального отсутствия реперфузии (феномен no-reflow).
2. Стадия транзиторной глобальной гиперемии развивается на 5–40-й минутах спонтанного кровообращения. Механизм ее развития связан с вазодилатацией сосудов головного мозга за счет повышения внутриклеточной концентрации Na+ и аденозина и снижения внутриклеточного рН и уровня Са2+. Длительность ишемии головного мозга в последующем определяет длительность стадии гиперемии, которая в свою очередь носит гетерогенный характер в различных регионах головного мозга, приводя к снижению перфузии и набуханию астроцитов.
3. Стадия пролонгированной глобальной и мультифокальной гипоперфузии развивается от 2 до 12 часов постреанимационного периода. Скорость церебрального метаболизма глюкозы снижается до 50 % от исходного уровня, однако глобальное потребление кислорода мозгом возвращается к нормальному (или более высокому) уровню в сравнении с исходным до момента остановки кровообращения. Церебральное венозное РО2 может находится на критически низком уровне (менее 20 мм рт. ст.), что отражает нарушение доставки и потребления кислорода. Причина этого заключается в развитии вазоспазма, отека, сладжирования эритроцитов и чрезмерной продукции эндотелиинов.
4. Данная стадия может развиваться по нескольким направлениям:
- Нормализация церебрального кровотока и потребления кислорода тканью мозга, с последующим восстановлением сознания. Сохранение персистирующей комы, когда как общий мозговой кровоток, так и потребление кислорода остается на низком уровне. Повторное развитие гиперемии головного мозга, ассоциированное со снижением потребления кислорода и развитием гибели нейронов.
В целом повышение внутричерепного давления (ВЧД) или снижение системного среднего артериального давления (САД) способно уменьшать церебральное перфузионное давление и таким образом вызывать серьезное повреждение нейронов. Патофизиологическими механизмами, обусловливающими персистирующую гипоперфузию головного мозга, являются:
- гипоксия или снижение АД; снижение деформируемости эритроцитов; повышение агрегационной активности тромбоцитов; развитие перикапиллярного клеточного отека; изменения концентрации Са2+.
В зависимости от уровня повреждения может наблюдаться различная степень угнетения сознания вплоть до атонической комы (кома III). Прогностически важным является состояние зрачков. Сохранение двустороннего расширения зрачков с изменением правильности их формы после прекращения действия «больших» доз адреномиметиков и холинолитиков свидетельствует о тяжелой степени энцефалопатии с неблагоприятным прогнозом для жизни. Даже в случае отсутствия подобной картины сохраняется вероятность необратимого повреждения корковых структур головного мозга с формированием к исходу острого периода неврологических признаков персистирующего, а затем и хронического вегетативного состояния.
Окончательный объем повреждения нервной ткани не определяется только эпизодом перенесенной гипоксии. Не менее опасно развитие реперфузионного синдрома, заключающегося в гиперемии на фоне утраты ауторегуляторной способности мозгового кровотока. Возобновление церебрального кровотока в варианте «роскошной» перфузии приводит к дополнительному повреждению клеток мозга за счет продуктов перикисного окисления, трансмембранным водно-электролитным и медиаторным дисбалансом. На органном уровне функциональная гиперемия сопровождается увеличением внутричерепного объема крови и повышением внутричерепного давления (ВЧД). Внутричерепная гипертензия представляет собой основной внутричерепной механизм прогрессирования повреждения мозга. В результате неконтролируемого увеличения внутричерепного объема крови, цитотоксического отека мозгового вещества складываются предпосылки для прогрессирования дислокационных механизмов, приводящих к вторичному повреждению подкорковых и стволовых отделов головного мозга. В конечной стадии наступает сдавление дренирующих вен (набухание головного мозга), критическое повышение ВЧД, затруднение церебральной перфузии вплоть до полного ее прекращения.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 |
