

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Метод очистки солей кипячением с малорастворимым или нерастворимым гидроксидом соответствующего металла заключается в том, что к раствору соли прибавляют некоторое количество взвеси гидроксида и раствор кипятят в течение 5—10 мин. После того как раствор остынет, гидроксид отфильтровывают, а раствор соли оставляют для кристаллизации.
Этот метод очистки основан на следующем. В растворе над осадком гидроксида создается определенное значение рН, что и вызывает осаждение соответствующих ионов. Например, если соли магния содержат соли железа, алюминия и марганца, то под влиянием гидроксида магния они будут осаждаться в виде гидроксидов. Степень очистки определяется произведением растворимости этих гидроксидов.
При равных значениях рН более растворимые гидроксиды будут осаждать менее растворимые. По убывающей растворимости все гидроксиды можно расположить в следующий ряд:
Mg(ОН) г, Ni(ОН)2, Со(ОН)2, Zn(ОН)2, Рb(ОН)2, Fе(ОН)2, Сu(ОН)2, А1(ОН)3 Fе(ОН)3
Метод наиболее пригоден для очистки солей магния, никеля и кобальта, так как гидроксиды этих металлов осаждают ионы большинства металлов.
Между растворимостью гидроксидов и значением рН, создаваемым ими, существует зависимость: хорошо растворимые гидроксиды создают более щелочную среду. Поэтому при очистке солей с помощью гидроксидов учитывают оба фактора. Теория очистки солей с помощью гидроксидов значительно сложнее, так как в большинстве случаев осаждаются не гидроксиды, а основные соли.
Гидроксиды необходимо готовить из тех солей, которые подвергаются очистке, или из более чистых и применять их в свежеприготовленном виде. После осаждения гидроксид следует тщательно промыть на фильтре теплой водой для полного удаления щелочи. Можно использовать для очистки и оксиды металлов, но только в тех случаях, когда они могут переходить, хотя бы частично, в гидратированную форму.
В отдельных случаях для очистки можно применять и другие вещества, например карбонаты и сульфаты.
4. Хроматографический метод
Хроматографический метод, предложенный в 1903 г. , основан на различной адсорбции веществ на слое сорбента. При движении раствора смеси двух веществ или смеси газов происходит многократная адсорбция и десорбция веществ, приводящая к тому, что в верхней части адсорбента, находящегося, например, в колонке, остается вещество, которое лучше адсорбируется; в нижней части — вещество, которое адсорбируется хуже. Если и дальше приливать в верхнюю часть колонки раствор смеси двух веществ, то из нее будет вытекать раствор хуже адсорбированного вещества. Можно также колонку с адсорбентом промыть раствором какого-либо комплексообразователя, дающего прочные комплексные соединения с одним из адсорбируемых веществ; в этом случае это вещество и будет вымываться из колонки.
Различные адсорбенты по-разному адсорбируют ионы. Например, на оксиде алюминия по способности адсорбироваться ионы можно расположить в следующий убывающий ряд:
![]()
![]() Fe3+ Co2+
Fe3+ Co2+
Cr3+ Al3+ > Zn2+ > Ni2+ Mn2+
Из приведенного ряда следует, что этим методом нельзя разделить ионы железы и хрома на оксиде алюминия. Но железо и хром будут сравнительно легко отделяться от марганца.
Существует несколько различных методов хроматографии: бумажная, газовая, пленочная и т. д. Для очистки солей наиболее приемлема колоночная хроматография с использованием катионообменных смол, содержащих в своем составе кислотные группы (—SО2ОН, —СООН, —SН и т. д.). При соприкосновении с растворами солей ион водорода замещается на катион, например:
R—SО2ОН + Сu2+ = (R—SО3)2Сu + 2H+
Аниониты содержат группы основного характера, например —NН2, дающие в растворе —NН3ОН. Группа —ОН может обмениваться на анионы.
Количество вещества, поглощенное ионитом, называется емкостью ионита.
Амфотерные иониты содержат и кислотные и основные группы и способны jсуществлять как катионный, так и анионный обмен. Для оценки эффективности ионита используют коэффициент разделения. Он представляет собой частное от деления отношений эквивалентных долей двух ионов в ионите и в растворе.
Коэффициент разделения — безразмерная величина и не зависит от выбора единиц концентрации веществ.
Лекция № 9-10. Реакции нуклеофильного замещения алкилгалогенидов.
Вопросы
Механизм реакций. Катализ. Соотношение между механизмами SN1 и SN2. Влияние строения реагентов на реакции нуклеофильного замещения. Конкуренция реакций нуклеофильного замещения. Правило Корнблюма.Реакции некаталитического нуклеофильного замещения могут протекать в зависимости от условий, характера реакционной среды, структуры субстрата и природы нуклеофильного реагента по одностадийному или двухстадийному механизмам.
Одностадийный процесс представляет собой элементарную реакцию SN2-замещения, где индекс 2 указывает на бимолекулярность реакции: 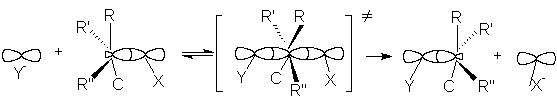
или более схематично
![]()
Можно видеть, что нуклеофил своей неподеленной парой электронов атакует углеродный атом в молекуле субстрата со стороны, противоположной замещаемой группе. При образовании активированного комплекса атакуемый атом углерода переходит из sp3-состояния в sp2-состояние. В результате углеродный атом оказывается в одной плоскости с группами R, R’ и R”, а нуклеофил и замещаемая группа связываются с ним за счет его р-орбитали. Распад активированного комплекса в продукт реакции сопровождается обратным переходом углеродного атома в sp3-состояние и обращением конфигурации исходного соединения, которое вызвано присоединением группы Y со стороны, противоположной разорвавшейся связи С-Х. Поэтому если объектом бимолекулярного нуклеофильного замещения является хиральный атом углерода, то в результате реакции происходит обращение оптической активности соединения. Скорость бимолекулярной реакции SN2 выражается уравнением
r = k[RX][Y-] (1)
При двухстадийном нуклеофильном замещении разрыв связи С-Х и образование связи С-Y происходит не одновременно. Первая элементарная стадия этой реакции - мономолекулярный гетеролитический разрыв связи С-Х с образованием карбкатиона
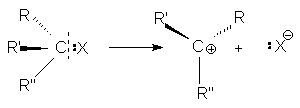
При этом центральный атом углерода переходит в состояние sp2-гибридизации и карбкатион приобретает плоское строение. Поэтому он с равной вероятностью атакуется нуклеофилом как со стороны отщепившейся группы, так и с противоположной стороны.
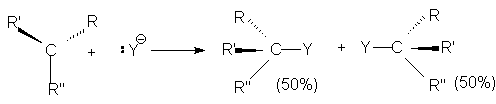
Поэтому, если объектом нуклеофильной атаки является хиральный атом углерода, то в результате реакции оптически активного субстрата образуется рацемическая смесь.
Первая стадия - мономолекулярная стадия гетеролиза - является скорость-определяющей. В связи с этим двухстадийный процесс нуклеофильного замещения обозначают символом SN1, где цифра 1 означает мономолекулярность скорость-определяющей стадии.
Скорость реакций SN1 в соответствии с характером скорость-определяющей стадии выражается уравнением
r = k[RX] (2)
В действительности механизм рассматриваемых реакций нуклеофильного замещения является более сложным.
В настоящее время общепринято, что разрыв связи С-Х существенно облегчается или вообще становится возможным лишь за счет взаимодействия замещаемой группы Х с электрофилами. Образование донорно-акцепторной связи I или водородной связи II
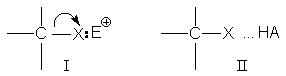
ведет к предварительной поляризации связи С-Х и облегчает ее последующий разрыв. Замещение некоторых групп (-OH, - OR, - SH, - SR, - NH2, - NHR, - NR2) вообще невозможно без их сильного взаимодействия с электрофилами. Например, замещение гидроксильной группы спирта при действии только бромид-аниона невозможно, но эту же реакцию легко осуществить в кислой среде. Предварительное протонирование гидроксильной группы меняет ее характер: в качестве уходящей группы выступает стабильная молекула воды, что делает энергетически выгодным гетеролиз связи С-ОН и облегчает атаку аниона брома
![]()
Аналогичным образом осуществляется превращение простых эфиров, меркантанов и аминов.
Кроме сильных протонных кислот (НСl, H2SO4) катализаторами реакций нуклеофильного замещения являются кислота Льюиса (Ag+, Hg2+, HgCl, HgCl2, SnCl4, AlCl3, SbCl5, Cu2Cl2, BF3 и др.). Их действие зависит от природы отщепляемой группы. Например, протонные кислоты существенно ускоряют замещение О-, S - и N-содержащих групп и мало влияют на скорость замещения галогенов, поскольку последние являются слабыми основаниями и плохо протонируются. Атомы галогенов дают, однако, прочные донорно-акцепторные комплексы с некоторыми кислотами Льюиса, и их образование облегчает разрыв связи С-Hal при последующей реакции замещения. Так, аллилхлорид медленно гидролизуется водой, но в присутствии хлорида меди (I) легко дает аллиловый спирт
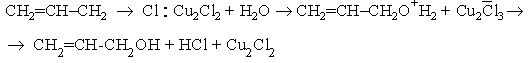
При этом может произойти изменение механизма SN2 в некаталитической реакции на SN1 - в каталитической:
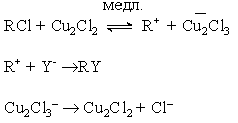
Ослабление связи С-Х и уменьшение энергии переходного состояния в реакциях нуклеофильного замещения происходит и при образовании водородных связей между уходящей группой и протонодонорным агентом.
Так, скорость реакции метанолиза трифенилхлорметана
(C6H5)3CCl + CH3OH → (C6H5)3COCH3 + HCl
описывается кинетическим уравнение третьего порядка:
r = k[(C6H5)CCl][CH3OH]2
Это согласуется с механизмом, согласно которому замещение хлора происходит только при условии его специфической сольватации другой молекулой спирта:
![]()

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 |
