
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Рис. 7.1. Временные зависимости молярных концентраций N исходных веществ А и продуктов реакции В и С В общем виде кинетическая модель системы из i компонентов с j независимыми химическими реакциями может быть описана m уравнениями через молярные концентрации N:
где |
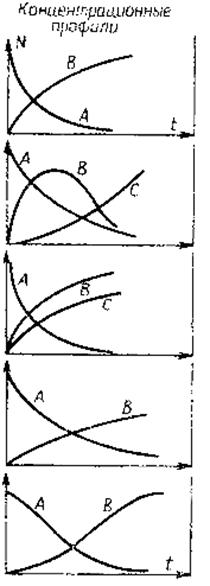
Таблица 7.1. Основные типы химических реакций и соответствующие им
временные зависимости молярных концентраций N исходных веществ А и продуктов реакции В и С
Тип реакций | Кинетическое уравнение | Кажущаяся константа скорости |
Реакция n-го порядка
| - |
|
Последовательные реакции
|
|
|
Параллельные реакции
|
| k1 +k2 |
Обратимая реакция
|
|
|
Автокаталитическая реакция
|
|
|
уравнений может быть решена методами минимизации, например итерациями по методу наименьших квадратов, естественно полагая, что число независимых уравнений всегда меньше числа констант скорости. При большем наборе экспериментальных данных интегрирование может быть выполнено численными методами. В случае ограниченного набора экспериментальных данных применяют специальные приемы, например сплайн-функции, для последовательного определения производных. Среди других методов решения следует упомянуть матричное преобразование Лапласа.
Не менее важной стадией большинства процессов является перенос вещества из одной части системы в другую либо в результате хаотичного движения (градиент концентраций и температуры), либо под действием внешних сил (электромагнитное поле, механические напряжения и т. д.). Процесс переноса вещества называют диффузией.
Подобно теплопроводности, стационарную диффузию можно описать первым законом Фика
![]() , (7.8)
, (7.8)
где J=(N/A) ‑ плотность потока вещества через площадь A, D ‑ коэффициент диффузии, ![]() ‑ градиент концентрации в направлении диффузионного потока х.
‑ градиент концентрации в направлении диффузионного потока х.
В случае нестационарной диффузии справедлив второй закон Фика
![]() (D = const). (7.9)
(D = const). (7.9)
Для решения этого уравнения, определяемого граничными условиями, на начальных стадиях процесса применяют функции ошибок, а на конечных стадиях ‑ тригонометрические ряды или функции Бесселя. Решение получают в виде графических зависимостей распределения концентрации во времени и пространстве (рис. 7.2). Из рис. 7.2 можно видеть, что решение диффузионных уравнений имеет вид зависимости от параметра![]() , что графически выражается параболой, а не прямой пропорциональной зависимостью, как в случае химических реакций. Аналогичный вывод получают и при решении уравнения первого закона Фика для стационарного роста слоя продукта на границе реакции, т. е.
, что графически выражается параболой, а не прямой пропорциональной зависимостью, как в случае химических реакций. Аналогичный вывод получают и при решении уравнения первого закона Фика для стационарного роста слоя продукта на границе реакции, т. е.
![]() , (7.10)
, (7.10)
где ![]() ‑ скорость роста слоя толщиной х и плотностью r при постоянном градиенте концентрации DN. При интегрировании получаем
‑ скорость роста слоя толщиной х и плотностью r при постоянном градиенте концентрации DN. При интегрировании получаем
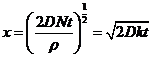 , (7.11)
, (7.11)
где К ‑ константа.
|
Рис. 7.2 ‑ Изменение концентрации N вещества в диффузионной зоне при различных временах диффузии ti (i =1- 4): Х ‑ пространственная координата, D ‑ коэффициент диффузии, d ‑ толщина; а ‑ диффузия из постоянного источника в бесконечный объем:N = 0 при x > 0 и t = 0 N = No при x = 0 и t > 0
б ‑ диффузия на границе двух бесконечных тел: N = 0 при x > 0 и t = 0; N = No при x < 0 и t = 0;
в ‑ диффузия из плоской бесконечной пластины, зависимость от толщины: N = No при - d > x £ d и t = 0; N = No при - d <x > d и t = 0;
|

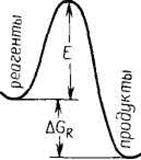
Любой из процессов предполагает транспорт реагирующих веществ из начального в конечное состояние, сопровождающийся преодолением определенного энергетического барьера, разделяющего эти два состояния. Одной из основных теорий современной химической кинетики является теория активированного комплекса. Эта теория предполагает существование некоего промежуточного реакционного состояния A# (рис. 7.3), которому можно приписать определенные термодинамические функции. В этом случае скорость процесса определяется скоростью распада комплекса А# до конечных продуктов. Математически скорость процесса выражают отношением kT/hK#, где k и h ‑ постоянные Планка и Больцмана, а К# имеет смысл константы равновесия, которая характеризует переход активированного комплекса в реагенты. Заменяя К#, получаем
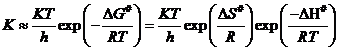 . (7.12)
. (7.12)
где DG# ‑ энергия Гиббса, DS# ‑ энтропия, DH# ‑ энтальпия процесса образования активированного комплекса из реагентов. Согласно теории активированного комплекса, процесс диффузии предполагает существование промежуточного состояния с наиболее высокой энергией, хотя вполне очевидно, что более выгоден и быстр путь с наименьшим энергетическим барьером (рис. 8.3, в).
|
|
Рис. 7.3 ‑ Графическое представление энергетического барьера процесса (Е) в соответствии с теорией активированного комплекса (отмечен *) для случая химической реакции (а), многостадийной (обычной каталитической) реакции с образованием промежуточного состояния (б) и для диффузии частиц из начального в конечное состояние (в) |
 а)
а)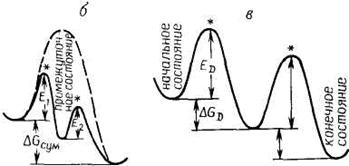
Многие теории предполагают установление взаимосвязи между параметрами уравнения (7.12) и измеряемыми величинами. При этом учитывают константы, характеризующие исходное состояние, геометрию и физические свойства образца. Определенные перспективы имеет гипотетическое приближение, использующее парциальные функции:
![]() ,
,
где Q# ‑ парциальная функция активированного комплекса (с ограниченной степенью свободы вдоль координаты реакции), Q ‑ парциальная функция реагента, Ео ‑ разность потенциальных энергий активированного комплекса и реагента, Zo ‑ предэкспоненциальный (частотный) фактор. Используя рассчитанные величины Q для процесса разложения СаСО3, найдены решения этих функций для характеристических колебаний решетки. При решении уравнения (7.13) для случая гипотетической гомогенной реакции исходят из предположения, что состояние молекул в твердой фазе можно описать

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 |
