
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
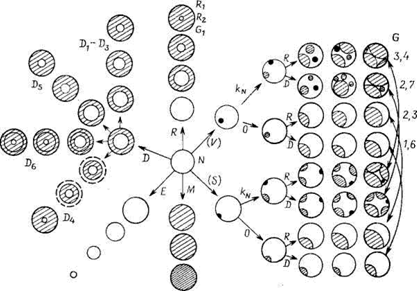
Модель сжимающейся реакционной границы принята при описании большинства реакций в твердых телах на основе геометрических представлений о реагирующей системе как наборе геометрических тел. Классификация и модификация всех индивидуальных типов процессов приведена на рис. 7.5. Ветви D и R схемы представляют два возможных пути протекания реакции с участием сферических частиц, которые с самого начала покрыты тонким слоем продукта, количество которого обычно экспериментально неопределимо. Реагирующие компоненты должны диффундировать через этот слой к (или от) реакционной границе, на которой протекает собственно химическая реакция. В этом случае скорость процесса определяется наиболее медленной стадией (диффузией и химической реакцией):
1) Если скорость процесса определяет химическая реакция, то имеет место реакция, контролируемая поверхностью (часто ошибочно называемая топохимической*). (*Согласно Освальду, процесс называется топотактическим, «если твердый продукт образуется в результате последовательности химических реакций или твердофазных превращений в одной или нескольких кристаллографически эквивалентных ориентациях по отношению к родительскому кристаллу и если он может заполнять весь объем родительского кристалла».)
|
Рис. 7.5 ‑ Представление основных типов гетерогенных процессов на основе модели сферической частицы: N ‑ зародышеобразование, D ‑ диффузия, R ‑ химическая реакция на поверхности, Е ‑ испарение, сублимация или растворение, М ‑ бездиффузионное фазовое выделение или мартенситное превращение без образования зародышей, G ‑ процесс, контролируемый ростом зародышей данного типа, происходящим благодаря диффузии (D) либо поверхностной реакции, S и V ‑ поверхность и объем, KN и 0 ‑ постоянная и нулевая скорости. |
Кинетическим параметром служит константа скорости k. Скорость реакции пропорциональна поверхности не прореагировавших частиц:
a = kA/V0, (7.20)
где Vo ‑ исходный объем частицы. Математическое преобразование приводит к уравнению
![]() , (7.21)
, (7.21)
в котором n=1/3, n =1/2 и n = 1 для случаев трехмерных, двухмерных и одномерных реагирующих частиц.
2) Если скорость процесса определяет массоперенос, т. е. диффузия, то главным процессом является постепенный рост слоя продукта, описываемый уравнениями (7.10) и (7.11). Яндер использовал параболический закон для описания реакций в порошковых системах
![]() . (7.22)
. (7.22)
Это уравнение, однако, справедливо только при многих упрощающих предпосылках (мгновенное поверхностное зародышеобразование, изотропная объемная диффузия, несмешиваемость реагентов и продуктов, исходные сферические частицы одинакового радиуса г0). Крегер и Циглер преобразовали это уравнение, введя предположение об обратной пропорциональности между D и t и заменив t на lnt:
![]() . (7.23)
. (7.23)
Журавлев дополнил уравнение зависимостью активности реагентов от доли не прореагировавшего материала (1-a):
![]() . (7.24)
. (7.24)
Гинстлинг и Броунштейн на основе выводов Баррера заменили параболический закон зависимостью, связывающей рост слоя продукта с площадью поверхности частицы. Тогда при стационарных условиях и сферической симметрии имеем
![]() . (7.25)
. (7.25)
Картер и Валенси ввели поправку на возможное изменение объемов начальной и конечной фаз во время процесса, используя соотношение
![]() , (7.26)
, (7.26)
где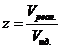 ‑ отношение идеального и реального объема растущего слоя. Хальберт вернулся к обратной пропорциональности между D и t и по аналогии с уравнением (7.23) заменил t на lnt в уравнении (7.26). Коматсу и Уемура ввели противодиффузию и получили так называемое антияндеровское уравнение
‑ отношение идеального и реального объема растущего слоя. Хальберт вернулся к обратной пропорциональности между D и t и по аналогии с уравнением (7.23) заменил t на lnt в уравнении (7.26). Коматсу и Уемура ввели противодиффузию и получили так называемое антияндеровское уравнение
![]() . (7.27)
. (7.27)
Сложный процесс, контролируемый множественной противодиффузией индивидуальных веществ А и В, может быть описан комбинацией допущений, предложенных Яндером и Коматсу и Уемурой. Если гА << rВ, то
![]()
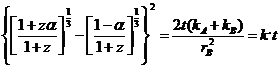 ,
,
где параметр z принимается равным отношению объема продукта, образованного на поверхности частицы В, к объему продукта, образованному внутри Аг и предполагается постоянным в течение всего процесса. Два предельных значения z сводят уравнение к более простым случаям: при z → ¥ к уравнению (7.27), при z → 0 к уравнению (7.22). Для реальных случаев, например для взаимодействия А12О3 с NiO, z = 3, что приводит к уравнению
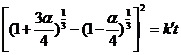 ,
,
тогда как при rа ~ rB получаем
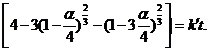
Затем уравнение (7.26) было модифицировано для случая полидисперсных систем с целью преодоления недостатков рассмотренных выше уравнений, поскольку они неправильно описывали конечные стадии порошковых реакций, на которых мелкие частицы полностью прореагировали. Очевидно, что допущения, принятые в уравнениях (7.22)-(7.27), можно комбинировать и дальше, а r0 может быть параметром, дополняющим а. В любом случае следует иметь в виду, что для процессов, контролируемых диффузией, в кинетические уравнения входит квадрат радиуса частиц, тогда как для процессов, контролируемых реакцией на поверхности, соблюдается прямая пропорциональность радиусу (го в первой степени). На практике очень трудно определить, какому закону подчиняется исследуемый процесс, в основном из-за полидисперсности частиц, нерегулярности их формы и значительной погрешности определения а.
Для случаев, когда покрытие поверхности частиц не является сплошным, Коматсу ввел в уравнение (7.22) корреляционные факторы, характеризующие процесс, который развивается не на всей поверхности частиц, а из ограниченного числа мест. Подобные процессы будут рассмотрены в следующем разделе. Влияние неравномерности смешения реагентов на возможность протекания параллельных процессов рассмотрено на рис. 7.6.

Рис. 7.6. Идеальный и действительный ход твердофазной реакции. Два твердых реагента А (светлые кружки) и В (заштрихованное поле) взаимодействуют в соответствии со схемой, давая конечный продукт АВ либо непосредственно, либо через образование промежуточных соединений А2В и А3В.
Образование этих промежуточных продуктов зависит, с одной стороны, от термодинамических и кинетических факторов, с другой — от локальной концентрации компонента А. В идеальном случае (/) реагент А равномерно распределен в соответствующем количестве реагента В. давая продукт АВ. Предположительно механизм реакции можно представить идеализированными кинетическими моделями, обсуждавшимися в предыдущих разделах (гл. 8 и 9, рис. 8.5). Для реальной смеси (2), когда компонент А распределен статистически, в областях, богатых реагентом А, может протекать реакция образования соединения АгВ, последующее разложение которого приведет к увеличению времени протекания суммарной реакции (а). В этом случае численная корректировка обычно удовлетворительно выравнивает неидеальность. Если компонент А образует агломераты (3), то при этом возможно образование всех термодинамически разрешенных продуктов, и общий ход реакции может протекать в осциллирующем режиме (б) из-за временного расхода конечного продукта АВ. Последний случай, однако, весьма сложен для выявления с помощью обычных термоаналитических измерений.
7.4. Процессы, контролируемые образованием и ростом зародышей. Кристаллизация
При выводе кинетических моделей зародышеобразования (рис. 7.5, ветвь N) предполагается, что скорость образования зародышей зависит от числа энергетически доступных мест No (в основном определяемых дефектами кристалла или примесями). Если в момент t число зародышей равно N, то для изотермического зародышеобразования справедливо уравнение
N = kN (N0 - N) (7.28)
Интегрирование и подстановка приводят к экспоненциальной зависимости
![]() (7.29)
(7.29)
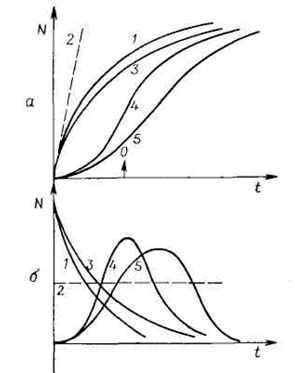
Если kN мало, уравнение (7.29) можно аппроксимировать линейной зависимостью N = kNN0t. Границы применимости уравнения (7.28) расширяются, если его дополнить либо степенной функцией вида (N0—N)n, что увеличивает или уменьшает наклон зависимости, либо множителем ехр(t/t), учитывающим возможную релаксацию (задержку) процесса зародышеобразования (рис. 7.7).
|
Рис. 7.7. Кинетические кривые зародышеобразования (а) и зависимости |
Образование зародышей продукта N и их последующий рост G можно описать в общем виде уравнением

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 |
