
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Таблица 7.2 ‑ ![]() Численные значения предэкспоненциального множителя для гомогенных твердофазных реакций первого порядка при 400 К*
Численные значения предэкспоненциального множителя для гомогенных твердофазных реакций первого порядка при 400 К*
Тип реакций | Предэкспоненциальный множитель Z0 | |||
Мономолекулярныереакции | Бимолекулярныереакции | |||
А | Б | А | Б | |
Без изменения вращательного состояния активированного комплекса и реагента | 1011 | 1015 | - | - |
То же, но с полностью ограниченным вращением активированного комплекса | 1011 | 1015 | 1012 | 1016 |
Активированный комплекс находится в более свободных условиях и вращается параллельно поверхности | 1012 | 1015 | 1014 | 1018 |
Реагент может свободно вращаться, в то время как активированный комплекс не может | 108 | 1012 | 1012 | 1010 |
Реагент находится в равновесии со слоем, адсорбированным на поверхности, из которого он превращается в продукт через активированный комплекс | - | 106 | - | - |
- - А – реакция на поверхности частиц размером 10 мкм, Б – разложение, протекающее в объеме твердой фазы.
функциями распределения молекул в газовой фазе, если не учитывать энергию сублимации. Рассчитанный таким образом набор значений Zo для различных условий представлен в таблица 7.2.
Далее необходимо также обсудить связь между термодинамическим (или статистическим) описанием данного процесса и экспериментально определяемой энергией активации. Эванс и Поляни пытались коррелировать Е с изменением энтальпии DН, которое в действительности является нижним пределом Е (рис. 7.3, б). Разность этих величин бывает как незначительной, так и очень большой, сравнимой с соответствующими величинами для гомогенных реакций (процессы разложения или кристаллизации стекла). Выводы о корреляции Е и DН послужили поводом для некоторых сомнений относительно правомерности применения закона Аррениуса к реакциям в твердой фазе (термически неактивируемые процессы) или в применимости теории активированного комплекса.
Проблема применимости закона Аррениуса хорошо сформулирована Витакером и Робинсоном: «Каждый верит в экспоненциальный закон распределения энергии, экспериментаторы ‑ потому, что они думают, что он может быть доказан математически, а математики ‑ потому, что верят, что он установлен экспериментально». Очевидно, что критиковать какой-либо кинетический метод, недостаточно точно характеризующий гетерогенный процесс, много легче, чем предложить более пригодную модель. Поэтому отказ от данного метода расчета кинетических параметров не выход из положения. Найти оптимальное компромиссное решение, позволяющее объяснить результаты измерений ‑ вот главная и самая трудная проблема в кинетике гетерогенных процессов, поскольку до сих пор не удалось предложить удовлетворительной математической модели реальных процессов. Поэтому в последующих разделах рассматриваются многие из известных феноменологических приемов получения численных значений кинетических параметров.
Более того, в твердофазных процессах появляется проблема формального отнесения величины энергии активации к одному молю, что в случае гетерогенных реакций, локализованных на границе раздела фаз, теряет свой первоначальный, установленный для гомогенных реакций смысл. Наиболее точно установлен физический смысл энергии активации для процессов диффузии, где энергия активации Е определена как сумма энтальпий образования DНОбр и движения DНдв. структурных дефектов.
Диффузионный механизм процессов в металлах может быть обусловлен разным характером движения дефектов. Это было показано на примере изучения диффузии в меди:
Тип движущегося дефекта | DНобр., КДж/моль | DНдв., Дж/моль | Е, кДж/моль |
Внедренные атомы Парный обмен Циклический обмен Вакансии Бивакансии Экспериментальные данные Экспериментальные данные | 820 - - 172 150 - | 12 985 370 94 37 - | 830 985 370 266 165-190 193 |
Как видно, экспериментальная величина Е хорошо согласуется с бивакансионным механизмом движения дефектов. Аналогичный анализ может быть полезен для интерпретации других экспериментальных величин Е.
Многие теории и экспериментальные работы посвящены коэффициенту диффузии. Поэтому полезно далее остановиться на этом параметре, хотя такое обсуждение довольно нехарактерно для формальной кинетики гетерогенных процессов. Знание подвижности атомов и ионов Вi может быть использовано, например, для определения скорости их движения под действием единичной силы. Предполагая, что движущей силой диффузии является градиент химического потенциала, имеем
![]() , (7.14)
, (7.14)
где Bi ‑ подвижность i-го компонента в диффузионном потоке Ji. Учитывая, что активность
![]() ,
,
получим
![]() . (7.15)
. (7.15)
Подстановка уравнения (7.15) в выражение первого закона Фика приводит к уравнению Нернста-Эйнштейна
Di = RTBi, (7.16)
которое особенно полезно при рассмотрении движения заряженных частиц и позволяет рассчитывать ионную проводимость кристаллов (и даже стекол):
![]() (7.17)
(7.17)
si ‑ проводимость, обусловленная транспортом частиц i-го сорта; Ni ‑ число ионов в 1 см3; Zi ‑ валентность; e ‑ заряд электрона; Bt ‑ абсолютная подвижность; s ‑ общая проводимость; t ‑ число переноса частиц i-ro сорта. Эти уравнения в ряде случаев бывают полезны при расчетах скорости диффузии из данных по проводимости.
В многокомпонентных системах коэффициент диффузии имеет сложный характер. Например, выравнивание концентраций в бинарных системах (сплавах) можно описать одним значением химического коэффициента диффузии D, являющегося функцией коэффициентов самодиффузии D компонентов А и В:
 . (7.18)
. (7.18)
В случае идеальных растворов (g = сonst) это уравнение преобразуется в тождество ![]() .
.
7.3. Физико-геометрическая модель процессов, учитывающая смещение межфазной реакционной границы от поверхности частицы к ее центру
Представим, что химическая реакция протекает на границе двух сред, т. е. образуется твердый продукт взаимодействия двух твердых реагентов. Кинетика реакций такого типа развита главным образом для плоских границ. Предполагают, что двухфазная система состоит из больших сферических частиц одной фазы, окруженных (покрытых) существенно более мелкими частицами другой фазы. Если есть уверенность, что поверхность больших частиц в существенной мере покрыта мелкими частицами (например, при их избытке) и все частицы находятся в плотном контакте (достигаемым, например, прессованием), то имеет место идеальный случай, в котором вместо мелкодисперсной фазы можно рассматривать жидкость. Такую систему можно описать на основе одной из следующих моделей:
1) нестационарная модель, предполагающая одновременное образование продукта реакции по всему объему крупных частиц;
|
Рис. 7.4 ‑ Схематическое представление временной зависимости образования частицы новой фазы (показана штриховкой) в первоначально пустой матрице исходной фазы в предположении скачкообразного (А) и непрерывного (В) изменения молярной концентрации N на поверхности: А ‑ пример стационарного процесса, характерного, например, для зародышеобразования (см. также рис. 5.4 и 8.7); В ‑ пример нестационарного процесса (фазовое выделение в спинодальной области, диффузионные (мартенситные) превращения и т. д.) |
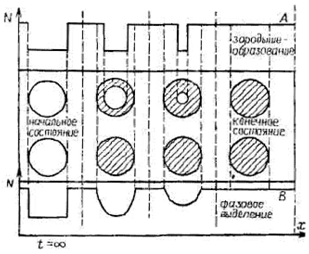
2) стационарная модель, описывающая постепенное образование продукта реакции за счет продвижения реакционной границы раздела (модель сжимающегося объема). Обе модели представлены на рис. 7.4.
Первая модель, предполагающая нестационарную объемную диффузию основывается на суммировании типа
![]() , (7.19)
, (7.19)
в котором k ‑ константа скорости ![]() , где г0 ‑ радиус реагирующих частиц, п ‑ целочисленная величина, характеризующая число членов бесконечного ряда, которыми ограничиваются при обработке зависимости a = f(t), D ‑ коэффициент диффузии). Первая модель одновременного объемного превращения удачно подходит для описания пористых материалов (например, таблетированных катализаторов) и особенно для описания технологических процессов. Технологические условия оцениваются в рамках обобщающего безразмерного параметра, называемого коэффициентом эффективности
, где г0 ‑ радиус реагирующих частиц, п ‑ целочисленная величина, характеризующая число членов бесконечного ряда, которыми ограничиваются при обработке зависимости a = f(t), D ‑ коэффициент диффузии). Первая модель одновременного объемного превращения удачно подходит для описания пористых материалов (например, таблетированных катализаторов) и особенно для описания технологических процессов. Технологические условия оцениваются в рамках обобщающего безразмерного параметра, называемого коэффициентом эффективности ![]() и определяемого отношением скорости реакции к единице поверхности реакционной зоны. Описание основано на предположении, что концентрация и температура в газовой фазе остаются независимыми от реакции в твердом теле. Излом на монотонных кривых зависимости h от а означает температурную нестабильность и изменение механизма реакции. Положительный наклон кривой
и определяемого отношением скорости реакции к единице поверхности реакционной зоны. Описание основано на предположении, что концентрация и температура в газовой фазе остаются независимыми от реакции в твердом теле. Излом на монотонных кривых зависимости h от а означает температурную нестабильность и изменение механизма реакции. Положительный наклон кривой ![]() определяет область геометрической нестабильности системы (в которой, например, модель усадки частиц становится неприемлемой для описания реакции из-за изменения реакционной поверхности).
определяет область геометрической нестабильности системы (в которой, например, модель усадки частиц становится неприемлемой для описания реакции из-за изменения реакционной поверхности).

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 |
