
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Метод (A): спів-осадження оксидів CeO2 і Sm2O3 з водних розчинів їх хлоридів. Попередньо отриманий водний розчин хлориду церію змішували з водним розчином хлориду самарію при температурі 40 ˚С та 500 об/хв. Після 30 хвилинної гомогенізації температуру підняли до 90 ˚С та збільшили оберти до 1000 об/хв. Попередньо отриманий розчин сечовини за допомогою пульверизатора додали до розчину. Отриманий осад промивали кілька разів у дистильованій і деіонізованій воді, щоб видалити залишкові продукти реакції. Після чого промивали 3 рази етанолом виробництва Kanto Chemicals, Японія і чистоти 99,5%. Після кожної промивки етанолом суспензію центрифугували з метою відокремлення порошку. Кінцевий варіант суспензії повільно випарювали на магнітній мішалці при температурі 50 ˚С з наступною сушкою при температурі 90 ˚С в сушильній шафі.
Спів-осадження оксидів CeO2 і Gd2O3 з водних розчинів їх хлоридів (Метод А). Водний розчин хлориду церію нагрівали до температури 90 °С при перемішуванні 1000 об/хв. Попередньо нагрітий до 60 °С водяний розчин сечовини за допомогою пульверизатора додавали в розчин хлориду церію. В таких умовах синтез тривав 4 години. Після чого розчин охолоджували до температури 3 ˚С за допомогою льодяної бані і додавали розчин хлориду гадолінію. На цьому етапі перемішування проводили на максимальних обертах. Після 30 хв перемішування 100 мл водяного розчину сечовини додавали до загального розчину. Отримана суспензія нагрівали до 80 °С і змішували протягом 5 годин. Отриманий осад промивали і сушили так само як і у випадку синтезу Ce0,8Sm0,2O2-δ.
Метод (Б): синтез горінням. Вихідні реагенти CeCl3·7H2O, SmCl3·6H2O та GdCl3·6H2O були зважені і змішані відповідно до складу твердих розчинів Ce0,8Sm0,2O2-δ та Ce0,8Gd0,2O2-δ. Отримана суміш (гель) була внесена в попередньо розігріту до 600 ˚С піч і витримана протягом 2 год.
Метод (В): водні розчини хлоридів церію, самарію та сечовини взяті у потрібному співвідношенні заливали в тефлоновий реактор і піддавали гідротермальної обробці при температурі 110 °С протягом 24 год. Отриманий осад промивали і сушили так само як і у випадку синтезу У випадку синтезу церій-самірієвого оксиду, оксид гадолінію синтезувався окремо в гідротермальних умовах при температурі 110 °С протягом 7 год. Отриманий осад промивали три рази в етиловому сирті. Синтезований порошок оксиду гадолінію додавали до розчину хлориду церію та сечовини і піддавали гідротермальній обробці протягом 24 год. Отриманий в результаті композит промивали три рази в етиловому спирті та піддавали термічній обробці.
Для визначення оптимальної температури термічної обробки сирі порошки піддавалися термо-гравиметричному аналізу (ТГ) на приладі Thermo Plus 2 Рігаку, TG8120, Рігаку, Японія. Al2O3 був взятий як еталонний зразок. Швидкість нагріву становила 5 ˚С/хв.
За результатами ТГ аналізу температуру 600 ˚С та витримку 2 години було обрано для неізотермічної обробки.
2.3 Приготування сумішей композиту В6О-В4С
Синтезований B6O порошок змішували з різною кількістю наноструктурного порошку карбіду бору (В4С) виробництва Sinopharm Chemical Reagent Co. Ltd., Китай – 3, 5, 10, 20, 40, 60, 80 мас.%, в середовищі дистильованої води протягом 12 годин з наступним випарюванням на магнітній мішалці та сушці в сушильній шафі при температурі 170 С протягом 6 годин.
2.4 Ущільнення порошків методом ІПС
Ущільнення порошків методом іскро-плазмового спікання проводили на апараті Dr. Sinter Model SPS-1050, SPS-Syntex, Японія принципова схема якого представлена на рис. 2.2.
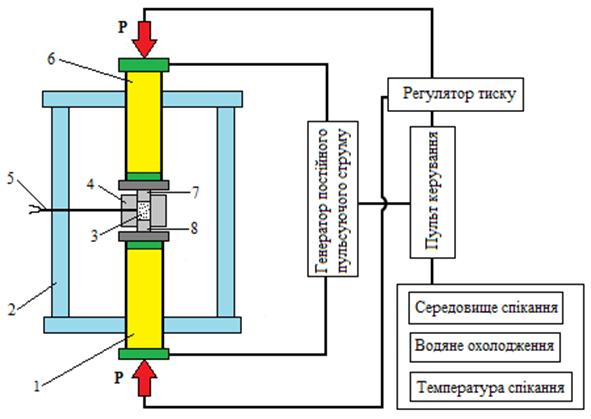
1 – нижній пуансон-електрод;
2– вакуумна камера із водяним охолодженням;
3– порошок;
4– графітова матриця;
5– термопара;
6 – верхній пуансон-електрод;
7, 8 – верхній та нижній графітові пуансони.
Рис. 2.2 – Принципова схема установки Dr. Sinter Model SPS-1050, SPS-Syntex, Японія
Для спікання методом ІПС використовували графітову матрицю (Toyo Tanso Co., Ltd., Японія) із зовнішнім діаметром 40 мм та внутрішнім діаметром 10 мм.. Зовнішня поверхня матриці була обгорнута графітовим войлоком товщиною 5 мм для зменшення втрати тепла за рахунок випромінювання. Внутрішня поверхня матриці покривалась графітовим папером для поліпшення контакту між матрицею та пуансонами, а також, щоб полегшити виймання зразка після спікання. Температура спікання контролювалася оптичним пірометром CHINO IR-AHS (Chino Co., Японія) з точністю 0,5 оС, який було направлено на матрицю з не наскрізним отвором глибиною 4 мм та діаметром 2,5 мм.
Температурно-часові режими спікання задавалися за допомогою блоку управління. Зовнішній тиск контролювали у ручному режимі. ІПС відбувалося при автоматичному контролю напруги та струму, максимальні значення яких сягали 4 В та 520 А, відповідно, при послідовності імпульсів 12:2 з тривалістю серії імпульсів 39,6 мс та міжсерійним інтервалом тривалістю 6,6 мс.
2.4.1 Ущільнення порошків В6О та В6О-В4С
У випадку спікання порошку В6О методом ІПС було застосовано три варіанти збору прес-форми: (1) між прес-формою та зразком був вставлений графітовий папір; (2) між прес-формою та зразком був вставлений графітовий папір з нанесеним на його поверхню hBN; (3) між прес-формою та зразком був вставлений графітовий папір, а між зразком і графітовим папером вставлялась танталової фольги виробнитства Sigma-Aldrich Chemie товщиною 0,025 мм і чистотою 99,9. Після аналізу отриманих результатів, тільки третій варіант використовувався для спікання композиту В6О-В4С. Спікання проводили у потоці аргону. Для встановлення залежності впливу тиску на властивості отриманого матеріалу, тиск змінювався в діапазоні 30–80 МПа. Тиск поступово збільшували при температурі більше 1700 °С від 10–15 МПа таким чином, щоб досягти потрібних значень (30–80 МПа) при завершенні нагріву до максимальної температури спікання 1800 °С. Зразки нагрівали до температури 1800 ˚С зі швидкістю нагріву 110 ˚С/хв, і витримували 1–10 хв. Кожен зразок поступово охолоджували до 600 ˚C зі швидкості охолодження 100 ˚С/хв, а далі зразки охолоджувались до кімнатної температури разом із піччю.
2.4.2 Ущільнення порошку Ce0,8Sm0,2O1,9
Порошок Ce0,8Sm0,2O1,9 спікали на повітрі при температурі 1000–1150 ˚С з витримкою 6–15 хв і максимальному тиску 150 МПа. Тиск поступово збільшувався від 10–15 МПа таким чином, щоб досягти 150 МПа при завершенні нагріву до максимальної температури спікання. Швидкість нагріву становила 400 ˚С/хв. Охолоджували зразок зі швидкістю 50 ˚С/хв. до 600 ˚С, потім разом із піччю до кімнатної температури. Зразки циліндричної форми з діаметром 10 мм товщиною близько 2 мм були отримані.
2.5 Реакційний синтез та синтез/спікання субоксиду бору в умовах ІПС
Для отримання В6О реакційним методом з використанням апарату ІПС суміш аВ і B2O3 Х14:1 (див п. 2.1) засипали в у графітову прес-форму і піддавали термічній обробці. Порошкову суміш нагрівали до температур 1200–1250 ˚С зі швидкістю 500 ˚С/хв і витримували протягом 15–30 хв. Мінімальний тиск 30 МПа було застосовано лише для досягнення контакту між зразком і прес-формою. Після синтезу пористий зразок подрібнювався.
Збір прес-форми для реакційного синтезу/спікання В6О був такий, як і для спікання, що описано в п. 2.4.1. Для попередження проникнення вуглецю в зразок була використана танталова фольга. Суміш Х14:1 нагрівали до температури 1250 ˚С (температури синтезу) зі швидкістю 500 ˚С/хв, і витримували 30 хв. Після цього температуру підвищують до 1600–1800 ˚С (температура спікання) зі швидкістю 110 ˚С/хв. Тиск поступово зростав до 80 МПа. Витримка становила 1 хв.
Синтезований порошок В6О в апараті ІПС був ущільнений при 1800 ˚С, витримці 1 хв та тиску 80 МПа.
Кожен зразок охолоджують до температури 600 ˚C зі швидкістю 100 ˚С/хв і далі охолоджували до кімнатної температури разом із піччю.
2.6 Традиційне спікання Ce0,8Sm0,2O1,9 на повітрі
Порошок Ce0,8Sm0,2O1,9 був попередньо спресований в зразок циліндричної форми діаметром 8 мм і висотою таблетки 4мм. Спресовані зразки завантажувалися в еластичну оболонку і піддавалися всебічному стиску в апараті холодного ізостатичного пресування (CL, Nikkiso Co., Ltd., Японія). при тиску 392 МПа протягом 10 хв. Отримані зразки спікалися на повітрі в печі резистивного нагріву при температурі ˚С з витримкою 1-10 годин. Всі зразки нагрівалися до температури спікання зі швидкістю 5 ˚С/хв, а охолодження відбувалося разом із піччю.
2.7 Методика рентгенографічних досліджень
Фазовий склад синтезованих порошків В6О та щільної кераміки на його основі досліджували методом рентгенівської дифрактометрії на приладі Rigaku Ultima IV, Japan в монохроматичному CuКα випромінюванні при напрузі 40 кВ і силі струму 40 мА. Дифрактограми знімали методом покрокового сканування в інтервалі кутів 2θ=10–90 град. Час експозиції в точці становив 4 с, крок сканування ― 0,02 град. Дифракційні картини одержували за кімнатних температур. Обробка дифрактограм проводилася за допомогою програмного забезпечення PDXL2 з вбудованими базами даних Міжнародного центру дифракційних даних (International Centre for Diffraction Data (ICSD)). Розмір ОКР був розрахований методом Вільямсона-Холу за допомогою програмного забезпечення PDXL2.
Фазовий склад порошків та спеченої кераміки Ce0,8Sm0,2O2-δ досліджували методом рентгенівської дифрактометрії на приладі Rigaku RINT 2000, Японія, в монохроматичному CuКα випромінюванні. Зйомка дифрактограм проводилася в діапазоні 2Θ =град з кроком 0,05 град з витримкою 5 сек при напрузі 40 кВ і силі струму 40 мА. Розшифровку дифрактограм проводили за допомогою комп’ютерної програми MDI Jade 5.0.
Метод повнопрофільного аналізу дифрактограм (метод Рітвельда) був використаний для розрахунку параметрів граток, координати атомів в кристалічних структурах та заповнення киснем позиції 6с у фазі В6О. Зйомку проводили на дифрактометрі Bruker AXS (D8 Advance, Німеччина) у CuKα випромінюванні при напрузі 40 кВ і силі струму 40 мА у інтервалі кутів 15-90 град. Крок сканування складав 0,02 ° з часом вибірки на кожну точку 10 с. Для обробки даних використовували комп’ютерну програму TOPAS
2.8 Методика дослідження морфології синтезованих порошків та мікроструктури спеченої кераміки
Дослідження морфології синтезованого порошку В6О методом трансмісійної електронної мікроскопії проводили на мікроскопі ПЭМ 125К (OAO «SELMI», Украина). Напруга прискорення становила 100 кВ.
Структурні дослідження синтезованого порошку В6О і композиційних матеріалів на його основі проводили методом скануючої електронної мікроскопії (СЕМ), на мікроскопі JEOL (JEM-2100F, Японія) з точковою роздільною здатністю 0,19 нм і напругою прискорення 200 кВ. Локальний рентгеноспектральний аналіз порошків проводили за допомогою мікроскопа SEM-EDX, Hitachi, Японія.
Морфологію синтезованих порошків Ce0,8Sm0,2O1,9 і Ce0,8Gd0,2O1,9 аналізували на трансмісійному електронному мікроскопі TEM (JEM-2000-FX, JEOL, Японія) з робочою напругою 200 кВ.
Дослідження мікроструктури і локальний розподіл по елементам спеченої кераміки Ce0,8Sm0,2O1,9 проводили на мікроскопах JEOL JEM-2100F і 7001F JEOL JSM (Японія). Розподіл елементів визначали за допомогою нанодисперсного рентгеноспектрального аналізатора (SEM-EDX)
Розподіл часток за розмірами було визначено шляхом випадкового вибору 200 частинок на фотографії за допомогою комп’ютерної програми SIAMS.
2.9 Методика вимірювання щільності спеченої кераміки
Густину всіх зразків спечених ІПС та традиційним методами вимірювали з використанням методу гідростатичного зважування (метод Архімеда). Розрахунки щільності зразків (ρзр)проводили по формулі (2.4).
![]() , (2.4)
, (2.4)
де М1 – маса зразка; М2 – маса зразка та води; М3 – маса зразка зануреного у дистильовану воду; ρ – густина дистильованої води (1 г/см3).
Для отримання маси М2 зразки занурювались у склянку з дистильованою водою і залишали на 24 години.
Зважування проводили на аналітичних вагах з дискретністю 0,0001 г.
Всі процедури зважування повторювали 5 разів і брали для розрахунків середнє значення. Теоретичне значення густини розраховували по формулі (2.5).
![]() , (2.5)
, (2.5)
де М – молярна маса; V – об’єм елементарної комірки; NAv – число Авогадро.
Теоретичну густину композитів визначали за рівнянням адитивності (2.6)
![]() , (2.6)
, (2.6)
де mi – масовий вміст компонента; ρi – теоретична щільність компонента.
2.10 Методика вимірювання мікротвердості і тріщиностійкості кераміки
Для вимірювання твердості та тріщиностійкості зразків використовували алмазний індентор Віккерса на приладі Micro Hardness Tester MHV1000 виробництва Китай. Навантаження на алмазну пірамідку було взято 9,8 Н. Час витримки під навантаженням складав 10 с. Щонайменше 10 вимірювання були проведені для розрахунку середнього значення твердості зразка.
Довжина тріщини після навантаження вимірювалася таким чином, як зображено на рис. 2.3.
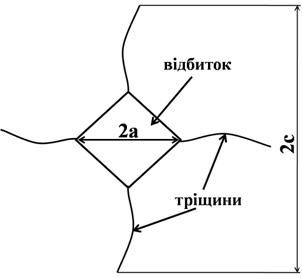
Рис. 2.3 – Схематичне зображення відбитка
Тріщиностійкість розраховували за рівнянням (2.7).
![]() (2.7)
(2.7)
де α – емпірична константа яка залежить від геометрії індентора; E – модуль Юнга, H твердість по Віккерсу, c – середній розмір тріщин; Р – навантаження на індентор.
2.11 Методика вимірювання іонної провідності
Електропровідність спечених електролітів Ce0,8Sm0,2O1,9 вимірювали за допомогою імпеданс спектроскопії [133]. Pt-паста наносилась з протилежних боків зразка. Потрібний контакт між пастою та зразком досягався шляхом відпалу при температурі 900 ˚С протягом 30 хв. Виміри проводили при температурі 300-800 ˚С з кроком 50 ˚С на повітрі. Для цього використовували імпеданс аналізатор Hewlett Packard HP 4194A з сигналом 50 мВ в діапазоні частот від 100 Гц до15 МГц. Еквівалентна схема (рис. 2.4) складалася з опорів R1, R2 та R3 і ємностей С1, С2 та С3.
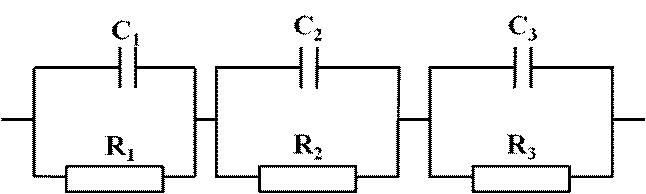
Рис. 2.4 – Еквівалентна схема виміру опору зразка.
За результатами проведених імпедансних вимірювань отримані частотні залежності перераховувалися в дійсну і уявну компоненти імпедансу і провідності, з наступним аналізом експериментальних даних в рамках наближення еквівалентної схеми. Відповідно до значень ємності, було встановлено що опір елемента R1 відповідає опору зерен (bulk), R2 опору границь зерен (grain boundary), R3 – опір електрода. Комплексний опір визначається як (2.8):
![]() (2.8)
(2.8)
де Z![]() - дійсна частина; Z
- дійсна частина; Z![]() - уявна частина
- уявна частина
Для еквівалента опору Z![]() =R1 Z
=R1 Z![]() =0, для еквівалента ємності Z
=0, для еквівалента ємності Z![]() =0;
=0;![]() , де ω – частота; С – ємність.
, де ω – частота; С – ємність.
Опір зерна матиме вигляд
![]() . (2.9)
. (2.9)
Опір границі зерна:

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 |
