


Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
II. Во-вторых, это произведение позволяет рассчитать любую из констант Kb и Ka, если известно значение другой из них.
11.2. Логарифмические показатели кислотности
1. а) Абсолютные значения вышеприведенных характеристик кислотности среды ([H+], [OH-]) и слабых электролитов (Ka, Kb) весьма малы: их можно
представить в виде 10-х, где обычно (хотя не всегда!) х > 1.
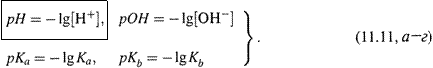 |
б) Поэтому чаще пользуются не самими этими характеристиками, а их логарифмами, взятыми с обратным знаком (т. е. величинами х из вышеприведенного представления 10-х):
(Точнее, в выражениях для рН и рОН должны фигурировать активности
соответствующих ионов, а не концентрации.)
Получим два ключевых соотношения для этих показателей.
2. а) Первое соотношение касается суммы рН и рОН. Оно исходит из уравнения диссоциации воды:
б) Константа равновесия этой реакции равна
в) Но диссоциирует очень малая часть воды; поэтому равновесная концентрация недиссоциированной воды практически равна общей концентрации воды:
Подставляя эту величину в (11.13), получаем значение ионного произведения
воды:
г) И, наконец, переходя к логарифмической форме, находим:
3. а) Это соотношение справедливо не только для чистой воды, но и для раствора любого электролита. Действительно, оно следует просто из константы KH2O, которая не зависит от присутствия других веществ.
б) Но для чистой воды, согласно уравнению ее диссоциации (11.12, б),
в) Заметим: из (11.16) вовсе не следует, что возможные значения рН и рОН заключены в пределах от 0 до 14. Так, пусть с(НС1) = 3 М. Соляная кислота —
сильный электролит, т. е. диссоциирует полностью. Известно, что при данной
концентрации коэффициент активности равен γ(НС1) = 1,3. Отсюда
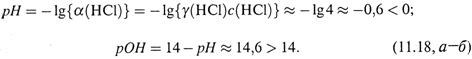 |
4. а) Получим второе соотношение, связывающее логарифмические показатели кислотности. Для этого достаточно прологарифмировать выражение, определяющее константу диссоциации слабой кислоты (11.7):
б) Как видим, ничего принципиально нового полученное уравнение не содержит. Это всего лишь иной вариант записи закона действующих масс для диссоциации слабого электролита. Используя его или изначальное выражение (11.7), рассмотрим следующие две ситуации.
I. Пусть слабый электролит находится в растворе с заданным рН. Тогда
уравнение (11.19,б) позволяет найти соотношение двух форм электролита
([R]/[RH]), а отсюда — и долю диссоциированных молекул (степень диссо-
циации).
II. Если же речь идет о чистом растворе слабого электролита, то, исходя из
(11.19, б), можно оценить рН, создающийся в этом растворе.
11.3. Влияние заданного рН на степень диссоциации
слабого электролита
Итак, обращаемся вначале к первой из указанных ситуаций.
1. а) Дадим определение: степень диссоциации α — это доля молекул в состоянии сопряжённого основания:
б) Тогда для слабой кислоты величина α — это действительно степень диссоциации, т. е. доля диссоциированных молекул.
в) А для слабого основания, при диссоциации которого отщепляется гидроксил–ион, доля диссоциированных молекул (т. е. молекул в состоянии сопряженной кислоты), очевидно, равна 1 — α. Выразим из уравнения (11.19,6) [RH] (концентрацию сопряженной кислоты):
 |
Подставляя в определение α (11.20, б), получаем:
Данная формула позволяет рассчитывать степень диссоциации слабого электролита (в соответствии с нашим определением) при произвольно заданном рН.
б) То же самое можно записать и в концентрационной форме. Действительно, выразим [RH] из (11.7) и вновь подставим результат в (11.20,б). Последовательно получим:
в) Нетрудно убедиться в тождественности выражений (11.22) и (11.23,б). Но первое из них предпочтительнее.
3. а) Подставим в формулу (11.22) три значения рН:
рН = рКа – 1,5, α ≈ 0,03 ,
рН = рКа, α = 0,5 ,
рН = рКа + 1,5, α ≈ 0,97 ,
и построим по этим точкам график зависимости α от рН (рис.11.1).
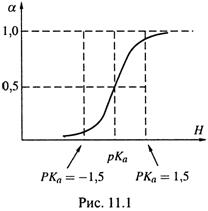 б) Прежде всего отметим: как показано на графике, степень диссоциации слабого электролита может меняться от 0 до 1.
б) Прежде всего отметим: как показано на графике, степень диссоциации слабого электролита может меняться от 0 до 1.
Это важно подчеркнуть потому, что нередко слабые электролиты определяют как вещества, степень диссоциации которых не превышает 3 % (0,03). Такое определение может относиться только к чистым растворам этих веществ (и то – лишь при достаточно большой концентрации). В общем же случае оно неверно.
в) Помещая слабый электролит в среду (например, буферную) с заданным значением рН, можно получать, согласно рис. 11.1, и гораздо большую степень диссоциации — практически вплоть до 1,0.
А изменение рН среды (например, с помощью сильных кислот и оснований) позволяет последовательно изменять и степень диссоциации слабого электролита.
г) При этом почти весь процесс диссоциации происходит в интервале рKa±1,5, т. е. от рKa—1,5 (где диссоциировано 3 % частиц) до рKa +1,5 (диссоциировано 97% частиц).
д) Центр этого интервала — такое значение рН, которое равно рKa — характеристике конкретного электролита. При данном рН диссоциировано ровно 50% частиц электролита.
11.4. Чистые растворы слабых кислот и оснований
Теперь обратимся ко второй ситуации, указанной в конце п.11.2. Будем считать, что в растворе – только слабый электролит. Найдём рН раствора и степень диссоциации электролита.
1. Закон разведения Оствальда. а) Для чистого раствора слабого электролита, диссоциирующего по уравнению
справедливо:
где с0 = [RH]0 — общая концентрация электролита.
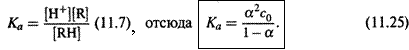 |
б) Вспоминаем выражение для константы кислотности:
Полученное выражение связывает степень диссоциации слабого электролита с общей его концентрацией. Оно-то и называется законом разведения Оствальда. Чтобы уяснить смысл этого закона, рассмотрим различные виды слабых электролитов.
2. Слабые кислоты.
а) Для них в чистом состоянии (и при с0 » Ка) α « 1; поэтому в знаменателе формулы (11.25) величиной α можно пренебречь. Тогда
Получается, что повышение концентрации раствора приводит к уменьшению степени диссоциации, а разбавление (или разведение) сопровождается увеличением диссоциации. Отсюда и термин – «закон разведения».
б) До какого уровня растёт α при разбавлении раствора? Выразив α из (11.25), можно показать, что
lim α = ,б)
со → 0
Таким образом, при очень сильном разбавлении (с0 « Ка) слабый электролит диссоциирует почти полностью.
в) Теперь, используя (11.24,а) и приближённую формулу (11.26,б), найдём концентрацию водородных ионов:
что в логарифмической форме выглядит так:
Заметим, что последнее выражение можно получить и непосредственно из формулы (11.19, б), если подставить в неё
г) Приведем пример использования этих выражений. Для уксусной кислоты рKa = 4,76. Пусть ее концентрация с0 = 0,1 М. Тогда по формуле (11.27, б) находим:
А чтобы воспользоваться формулой (11.26,б), учтем:
Отсюда
3. Слабые многоосновные кислоты.
а) Такие кислоты имеют несколько стадий диссоциации протона, например:
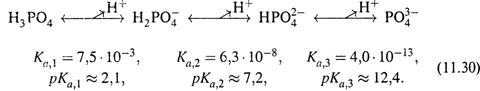 |
Каждая стадия характеризуется своей константой диссоциации; как видно из примера, константа каждой последующей стадии на несколько порядков меньше предыдущей.
б) Это значит, что в чистом растворе можно учитывать только первую стадию ионизации и рассчитывать рН раствора по формуле для одноосновной кислоты путем подстановки pKa, 1 в качестве pKa..
Так, для 0,1 М раствора H3PO4
4. Слабые основания.
а) Здесь α (доля молекул в состоянии сопряженного основания) близка к 1 (α ≈ 1); поэтому непосредственно использовать формулу закона Оствальда (11.25) неудобно.
б) В этом случае следует обратиться к уравнению вида
характеризуемому константой основности
и ввести степень диссоциации по этому уравнению:
в) В результате нетрудно получить формулы точно такого же вида, как для слабой кислоты:
г) Первая формула показывает, что степень оснóвной диссоциации тоже увеличивается при уменьшении с0, т. е. при разведении раствора. Закон Оствальда справедлив и здесь.
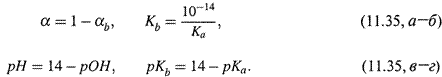 |
д) Но, как мы уже знаем, слабое основание можно характеризовать также константой кислотности и кислотной степенью диссоциации, причем
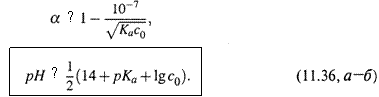 |
е) Подставляя первые два из этих выражений в (11.34, а), а два последние — в (11.34,б), для чистого раствора слабого основания находим :
ж) Пример — диссоциация аммиака
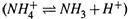 , pKa = 9,25. И пусть вновь с0 = 0,1 М. По формулам (11.36) получаем:
, pKa = 9,25. И пусть вновь с0 = 0,1 М. По формулам (11.36) получаем:
Таким образом, в чистом 0,1 М растворе аммиака основная доля частиц находится в состоянии сопряженного основания (NH3). Это справедливо и для всех других слабых оснований.
11.5. Чистые растворы солей слабых кислот и оснований
Итак, чистые растворы слабых кислот имеют слабокислую среду (рН < 7), а растворы слабых оснований – слабоосновную среду (pH > 7).
Для растворов же солей этих кислот и оснований приведём лишь конечные выражения расчёта рН, не останавливаясь на их выводе.
1. Предварительно следует заметить: в этих ситуациях рН среды изменяется в результате гидролиза соли. Поэтому вспомним: гидролиз соли — это взаимодействие её с водой, приводящее к образованию малодиссоциированпых соединений.
2. Соответствующие формулы представлены в таблице 11.1.
а) Как видно, первая из этих формул аналогична по виду формуле (11.36,б) для раствора слабого основания. Правда, теперь рКа относится к соответствующей слабой кислоте.
Т а б л и ц а 11.1
Растворённое вещество | Формула расчёта рН раствора |
Соль слабой кислоты и сильного основания | рН ≈ ½ (14 + рКа + lg cо ) (11.38) |
Соль сильной кислоты и слабого основания | рН ≈ ½ (рКа – lg cо) (11.39) |
и слабого основания | cо + Ка,1 рН ≈ ½ рКа,1 + рКа,2 + lg –––––––– (11.40) cо |
Но рассчитываемое значение рН оказывается больше 7. Это, в общем, естественно: преобладает влияние второго компонента соли — катиона сильного
основания.
б) Сходное замечание можно сделать по поводу второй формулы.
в) Пример: 0,1 М растворCH3COONa. Реакция гидролиза приводит к избытку гидроксил-ионов:
По формуле (11.38) получаем: рН ≈ 8,9.
Краткое содержание главы 11
Глава была посвящена растворам.
1. Мы вспомнили характеристики кислотности среды ([H+], [OH–]) и слабого
электролита (Ка, Кb), а также производные от них величины:
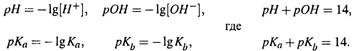 |
2. Соответственно, в двух формах может быть записан закон действующих масс для
диссоциации слабого электролита:
Затем были рассмотрены две ситуации.
3. Первая — ДИССОЦИАЦИЯ СЛАБОГО ЭЛЕКТРОЛИТА В СРЕДЕ С ЗАДАННЫМ рН. Степень диссоциации (определенная как доля частиц в состоянии СОПРЯЖЕННОГО ОСНОВАНИЯ) вычисляется так:
В зависимости от рН, она может изменяться от 0 до 1.
4. Вторая ситуация — ЧИСТЫЕ РАСТВОРЫ слабых электролитов. Для нее, в част-
ности, получены формулы расчета степени кислотной диссоциации и рН среды:
![]()
СЛАБЫЕ КИСЛОТЫ:
![]()
СЛАБЫЕ ОСНОВАНИЯ:
![]() СОЛЬ СЛАБОЙ КИСЛОТЫ
СОЛЬ СЛАБОЙ КИСЛОТЫ
И СЛАБОГО ОСНОВАНИЯ:
Глава 12. БУФЕРНЫЕ СИСТЕМЫ
Итак, в отношении слабых электролитов имеют место два факта:
1) чистые растворы таких электролитов изменяют рН растворителя (воды)
в ту или иную сторону,
2) изменение рН среды с помощью сильных щелочей и кислот приводит к изменению степени диссоциации слабого электролита — от 0 до 1 (или наоборот).
Особое значение имеет второй факт: благодаря ему растворы слабых электролитов обладают буферными свойствами. Эти свойства мы и обсудим в данной главе.
12.1. Буферное действие растворов слабых электролитов
1. а) Для понимания сути буферного действия рассмотрим построенный на
рис. 12.1 график титрования слабой кислоты сильной щелочью. Заметим, в качестве слабой кислоты можно рассматривать любую сопряженную слабую кислоту, в том числе, например, ![]() и т. д.
и т. д.
б) На таких графиках по горизонтальной оси обычно откладывают рН, а по вертикальной оси — молярную концентрацию эквивалента добавляемой щелочи в рассматриваемом растворе, обозначенную с(В).
в) Не следует забывать, что здесь первичным является добавление щелочи, а вторичным — изменение (в связи с этим) рН.
2. Итак, допустим, что имеется чистый раствор слабой кислоты с pH0,, к которому добавляется щелочь, т. е. увеличивается с(В).
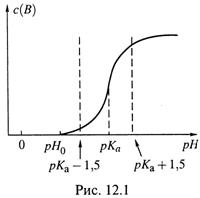 а) Пока не будет достигнут рН и рКа — 1,5, слабая кислота практически вся находится в недиссоциированном состоянии и не влияет на рН. Поэтому до
а) Пока не будет достигнут рН и рКа — 1,5, слабая кислота практически вся находится в недиссоциированном состоянии и не влияет на рН. Поэтому до
данного уровня изменение рН происходит быстро.
б) Но по достижении указанного значения рН начинается заметная диссоциация слабой кислоты:
Образующиеся протоны будут связывать бóльшую часть добавляемых гидроксильных ионов, и изменение рН станет гораздо более медленным, хотя
совсем не прекратится.
в) В этом-то и состоит буферное действие слабых электролитов: их присутствие препятствует резкому изменению рН при добавлении щелочных или
кислотных соединений, а при непрерывном добавлении последних — снижает
скорость изменения рН.
Зона же рКа±1,5 называется зоной буферного действия слабого электролита. (Иногда за таковую принимают более узкий интервал — рКа± 1.) Продолжающееся добавление щелочи, хотя и медленно, но повышает рН среды.
г) Когда рН выходит за верхнюю границу буферной зоны, буферное действие
прекращается. К этому времени практически весь слабый электролит переходит в форму сопряженного основания (R) и перестает реагировать на дальнейшее повышение рН, в связи с чем рН вновь начинает быстро возрастать.
3. а) Если теперь (после достижения высокого уровня рН) начать добавлять
сильную кислоту, то все будет происходить в почти обратном порядке. «Почти» — оттого, что вначале некоторое количество кислоты потребуется на нейтрализацию избытка щелочи.
б) Затем (при дальнейшем добавлении кислоты) вначале будет наблюдаться
быстрое снижение рН, затем (в зоне буферного действия слабой кислоты) —
медленное, а после выхода из этой зоны — опять быстрое понижение рН.
12.2. Расчет рН раствора с буферной системой
1. а) Заметим: в зоне буферного действия слабой кислоты в растворе присутствуют два компонента — сама кислота и ее соль с сильным основанием:
![]() RH ↔ R + H+
RH ↔ R + H+
сопр. к-та сопр. осн. . (12.1,a-б)
BR → B+ + R
б) Поэтому, если хотят составить уже готовую буферную систему, в раствор вносят два вещества:
I. либо, как в (12.1), слабую кислоту и её соль с сильным основанием,
II. либо слабое основание и его соль с сильной кислотой.
2. Поясним ещё раз, почему необходима соль.
а) В её отсутствие имелся бы чистый раствор слабого электролита, а в таком растворе, как было видно из оценок (11.29,а-б), степень диссоциации очень низка и рН находится вне зоны буферного действия (точка рН0 на рис.12.1). Поэтому при внесении в раствор, например, сильной кислоты получился бы сильный скачок в кислую сторону.
б) В присутствии же соответствующей соли сразу устанавливается рН, находящийся в пределах буферной зоны.
3. а) Чтобы рассчитать это значение рН, будем исходить из уравнения:
которое связывает рН с равновесными концентрациями двух форм слабого
электролита.
б) Обратимся к системе (12.1). В первом уравнении данной системы в чистом
растворе слабой кислоты равновесие резко сдвинуто влево, а в присутствии
соли, т. е. дополнительных частиц R, оно еще более сдвигается влево. Поэтому
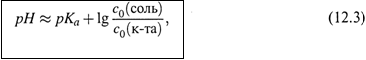 |
где с0(соль) и с0(кислота) — общие концентрации компонентов буферной
системы. В итоге для буферной системы получаем уравнение Гендерсона—
Хассельбаха:
которое в отличие от формулы (11.19, б) является приближенным.
в) Таким образом, значение рН буферной системы определяется отношением
концентраций соли и слабой кислоты. А поскольку при разбавлении раствора
это отношение не меняется, то не меняется при разбавлении и рН буферной системы. Это отличает ее от чистого раствора слабого электролита, для которого справедлив закон разведения Оствальда (п. 11.4).
4. а) Если речь идет о буферной системе типа II (слабое основание и его соль с сильной кислотой), то химические уравнения таковы:
 ВОH ↔ В + ОH–
ВОH ↔ В + ОH–
сопр. осн. сопр. к-та (12.4)
BА → B + А–
б) Уравнение (11.19,б) принимает вид:
причем
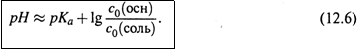 |
в) Это дает следующее уравнение Гендерсона-Хассельбаха:
5. Пример – ацетатный буфер, т. е. смесь уксусной кислоты (рКа = 4,76) и ацетата Na.
а) Допустим, нужен раствор с рН = 5,0. Тогда из уравнения (12.3) находим:
В таком соотношении для получения заданного значения рН надо смешивать компоненты системы.
б) Теперь пусть приготовлен буфер именно с указанным отношением концентраций, причем конкретные значения последних таковы:
К раствору добавлен NaOH до с(В) = 0,01 М.
I. В отсутствие буфера это привело бы к резкому увеличению рН среды:
Таким образом, если бы изначально была чистая вода (рН = 7,0), то рН увеличился бы на 5 единиц.
 |
II. Наличие же буферной системы приводит к следующему результату: добавление NaOH стимулирует диссоциацию соответствующего количества слабой кислоты, так что концентрация соли в буферной системе возрастает, а концентрация кислоты — уменьшается:
Отсюда новое значение рН равно:
т. е. по сравнению с исходным значением (5,0) рН изменился всего на шесть сотых единицы. Это — наглядная демонстрация буферного действия.
12.3. Буферные свойства многоосновных кислот
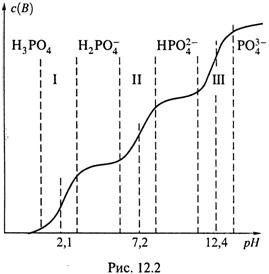
Рассмотрим кратко буферные свойства фосфорной кислоты.
1. График её титрования (рис. 12.2) включает не одну, а три буферные зоны с центрами в точках рКа,1, рКа,2 и рКа,3, довольно далеко отстоящих друг от друга.
 |
а) Вне буферных зон кислота находится, в основном, в одной из своих
четырех возможных форм:
б) В пределах каждой зоны сосуществуют две смежные формы, и соотношение между ними меняется по мере изменения рН.
Это значит, что в каждой такой паре форм одна выступает в качестве сопряженной кислоты, а вторая — в качестве сопряженного основания, поэтому к любой такой паре применимы полученные выше соотношения.
2. Так, например, если мы хотим получить буферный раствор с рН в пределах второй буферной зоны, нам надо взять соли (калиевые или натриевые) двух следующих анионов — ![]() . Первый из этих анионов выступает как кислота:
. Первый из этих анионов выступает как кислота:
 |
поэтому формулу Гендерсона—Хассельбаха в данном случае следует записать так:
Заметим: такой простой подход возможен лишь тогда, когда буферные зоны не перекрываются. В противном случае расчет является более сложным, т. к. при рН, находящемся в пределах области перекрывания, сосуществуют не две, а больше различных форм вещества.
12.4. Буферная сила и буферная емкость
а) Пусть имеется буферная система и к ней добавляется сильная кислота или
щелочь. Сколько надо кислоты или щелочи, чтобы изменить рН, скажем, на одну единицу?
б) Очевидно, чем больше требуется при этом кислоты или щелочи, тем эффективней буферная система. В связи с этим, известны две характеристики буферных систем.
1. а) Буферная сила — это дифференциальная характеристика:
Здесь KB — буферная сила по сильному основанию, а KA — буферная сила по кислоте.
б) Фактически, каждая из данных величин — это производная (с положительным или отрицательным знаком) кривой титрования, если в качестве аргумента брать рН, а в качестве функции — с(В).
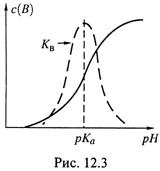 в) Поэтому зависимость KB от рН имеет колоколообразный характер (рис. 12.3) с максимумом в центре буферной зоны (где скорость dC(B)/d(pH) максимальна). При удалении от центра буферная сила быстро убывает и вне буферной зоны практически сводится к нулю.
в) Поэтому зависимость KB от рН имеет колоколообразный характер (рис. 12.3) с максимумом в центре буферной зоны (где скорость dC(B)/d(pH) максимальна). При удалении от центра буферная сила быстро убывает и вне буферной зоны практически сводится к нулю.
г) Абсолютная величина буферной силы зависит от общей концентрации компонентов буферной систе-
мы. Так, можно убедиться, что в центре буферной
зоны
Значит, чем больше концентрация буферной системы, тем больше требуется щелочи для одного и того же изменения рН.
д) Естественно также, что буферная сила по основанию и по кислоте при любом рН одинакова, но различна по знаку.
2. а) Однако на практике буферную систему характеризуют отношением не дифференциалов, а конечных приращений концентраций щелочи (кислоты) и рН.
Это отношение называют буферной ёмкостью — основной (В) или кислот-
ной (А):
б) Таким образом, В показывает, на сколько требуется увеличить в среде
концентрацию щелочи для увеличения рН на 1.
в) Если приращения Δc(B) и Δ(pH) невелики, то понятие буферной емкости практически совпадает с понятием буферной силы. И, соответственно, для В справедливо то, что относилось к буферной силе: колоколообразный характер зависимости от рН, а также зависимость от общей концентрации компонентов буферной системы.
3. а) Но можно задаться тем же вопросом буквально: на сколько требуется увеличить концентрацию щелочи в среде, чтобы рН раствора реально повысился
на 1 с какого-то начального значения рН0?
б) Для исходного значения рН имеем:
При увеличении же концентрации щёлочи на искомую величину В уравнение приобретает вид:
в) Вычитая из этого равенства равенство (12.6), можно найти:
9 с0 (к-та) × с0 (соль) 9 с0 (к-та) × с0 (соль)
В = –––––––––––––––––– и (аналогично) А = –––––––––––––––––––. (12.19,)
с0 (к-та) + 10 с0 (соль) 10 с0 (к-та) + с0 (соль) а-б)

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 |
