


Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Итак, плавление льда примерно так же увеличивает энтропию, как последующее нагревание воды на 100 градусов (3.15,б).
Пример 2. Аналогично можно найти изменение энтропии в результате испарения воды при температуре кипения (Ткип = 373 К).
Теплота испарения:
Отсюда |
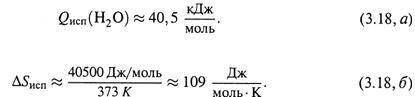
4. Абсолютная энтропия; энтропии образования и сгорания.
а) Как видно, часто достаточно оперировать изменениями энтропии, а не самими ее абсолютными значениями.
Тем не менее, исходя из третьего начала термодинамики (3.10), можно рассчитывать и абсолютную энтропию вещества или какой-то более сложной системы при некоей температуре Тх.
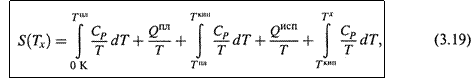 |
б) Пусть Тх выше температуры кипения. Тогда справедливо следующее выражение:
где учтены три этапа нагревания вещества и два фазовых перехода между ними.
В общем случае теплоёмкость, фигурирующая в (3.19), зависит от температуры. Поэтому её определяют в калориметре при разных температурах, строят график функции CP/T для каждого этапа и измеряют площадь под кривой (т. е. проводят интегрирование графическим способом).
в) В таблицах обычно приводят абсолютную энтропию (S0), относящуюся к стандартным условиям (1.3):
Т = 25° (298 К), Р=1 атм (101,3 кПа), п = 1 моль.
Например, для воды расчет по формуле (3.19) дает:
а для молекул пара воды при той же температуре, учитывая (3.18, б) и пренебрегая зависимостью ΔSисп от Т, находим:
г) Кроме того, как и для энтальпии (п. 2.3), употребляются следующие понятия:
ΔS0обр — стандартная энтропия образования вещества X, т. е. изменение энтропии системы в реакции (2.14, а):
простые вещества → X;
ΔS0сг — стандартная энтропия сгорания вещества, относящаяся к реакции (2.14,б):
Х + О2 → оксиды (с макс. СО).
5. Изотермическое расширение и сжатие газа.
а) Если допустить, что процесс идет обратимо, и использовать формулы (1.8) и (1.6):
то изменение энтропии определяется выражением:
Пример. При расширении газа в 2 раза (т. е., если Р1/P2 = 2), в расчете на 1 моль,
б) Но и при необратимом способе расширения изменение энтропии будет точно таким же! Это следует из того, что энтропия — функция состояния. Следовательно, формула (3.21,б) справедлива, независимо от способа расширения (или сжатия) газа. В частности, ею можно пользоваться при расчете ΔS в следующих двух ситуациях, эквивалентных расширению газа в пустоту.
I... Газ занимает объем V1, а ему дают возможность занять больший объем
V2 . Если в дополнительном объеме (V2 – V1) вначале газа не было (т. е. Р = 0), то при расширении газ не совершает никакой работы. Но энтропия его увеличивается, согласно формуле (3.21,б).
II. Аналогичная ситуация наблюдается при смешении разных газов.
6. Изотермическое изменение концентрации раствора.
а) Для газов давление и концентрация прямо связаны друг с другом. Действительно, из уравнения Клайперона-Менделеева
следует:
Поэтому, вновь исходя из (3.21,б), для изменения энтропии при изменении концентрации газа от с1 до с2 получаем формулу:
б) Разбавленные растворы во многом подобны газам. Во всяком случае, энергетический беспорядок системы (т. е. энтропия) при разбавлении или концентрировании растворённого вещества должен меняться точно так же, как при сгущении или разрежении газа.
Следовательно, и здесь ΔS рассчитывается по формуле (3.24).
В частности, если с2 < с1 (концентрация уменьшается), то ΔS > 0: энтропия (в расчете на моль вещества) возрастает, так как молекулы получают возможность располагаться более произвольно. Причем при разбавлении вещество не совершает работы, поскольку давление системы при этом не меняется, т. е. процесс эквивалентен расширению газа в пустоту.
Таким образом, изменение концентрации растворенных веществ (для достаточно разбавленных растворов и при постоянной температуре) не сопровождается поглощением или выделением теплоты (в том случае, конечно, когда вещество не взаимодействует с растворителем).
3.5. Статистическая природа энтропии
Конкретизируем представление об энтропии как о мере энергетического беспорядка.
1. Для этого введём два понятия – макро - и микросостояния системы:
а) макросостояние характеризуется макропараметрами – температурой, давлением, объемом, внутренней энергией и т. д.;
б) микросостояние — это конкретное расположение отдельных частиц (молекул) в данный момент времени, а также их скорости и взаимодействия.
Очевидно, что одно и то же макросостояние может реализоваться множеством различных микросостояний. Поэтому для каждого микросостояния существует определенная вероятность pi того, что система находится именно в данном микросостоянии.
Ясно, что S pi = 1.
2. а) Статистическая физика доказывает, что энтропия с точностью до постоянного множителя — это средний логарифм вероятностей микросостояний (возможных при данном макросостоянии), взятый с обратным знаком:
 |
где Ω — общее число возможных микросостояний.
б) Согласно приведенной формуле, величину беспорядка в системе определяют два фактора.
I. Первый – число микросостояний (Ω): чем оно выше, тем больше членов в сумме (3.25) и, следовательно, меньше средняя вероятность, приходящаяся на один член. Ясно, что при этом выше и беспорядок в системе.
II. Второй фактор — то, насколько микросостояния различаются по своей вероятности pi: чем ближе микросостояния по вероятности, тем беспорядок больше. А если все pi одинаковы, то (при одном и том же Ω) энтропия максимальна.
3. а) Последний случай (равновероятность всех микросостояний: все pi = p) обычно реализуется для системы из очень большого числа частиц, и тогда остается влияние только одного фактора — числа микросостояний (Ω), которое теперь очень просто связано с вероятностью микросостояния:
б) Выполним для данного случая простые преобразования:
в) Наконец, заметим, что коэффициент А в формуле (3.25) — это константа Болъцмана:
имеющая, как видим, размерность энергии (отнесенной к температуре и одной частице). Следовательно, с ее помощью совершается переход от просто «беспорядка» системы к «энергетическому беспорядку».
г) В итоге, для системы с равновероятными микросостояниями получаем знаменитую формулу Больцмана:
 или, в расчете на 1 моль частиц,
или, в расчете на 1 моль частиц,
Как видно, абсолютная энтропия системы просто связана с числом возможных микросостояний.
д) Изменение же энтропии в процессе определяется изменением этого числа:
4. Для иллюстрации представления о микросостояниях обычно приводят следующий пример.
а) В системе — N молекул. Каждая из них характеризуется в некий момент времени координатами хi, yi, zi и энергией Еi.
Вместе эти 4 значения образуют точку в 4-мерном координатно-энергетическом пространстве.
 |
б) Данное пространство определенным образом разбивается на т ячеек. Если допустить, что каждая частица с равной вероятностью может находиться в любой из ячеек, тогда общее число микросостояний можно рассчитать по формуле:
Так, если N = 10 частиц, а m = 5 ячеек, то
5. Для оценки множества микросостояний реальной системы применим формулу Больцмана к воде. Используем оценки (3.20,а-б) абсолютной энтропии воды и пара (в Дж/моль·К) при 25° С:
Подстановка этих величин в (3.27,б) даёт: число микросостояний в расчёте на 1 моль частиц равно
Получается, что на 6·1023 частиц 1 моля жидкой воды при 25° С имеется меньше 5000 равновероятных микросостояний. В газообразном макросостоянии число микросостояний почти на 6 порядков выше.
3.6. Математическая формулировка второго начала
термодинамики
Согласно п. 3.1, термодинамическая осуществимость процесса определяется балансом двух критериев: теплоты процесса и изменения энтропии.
1. а) С учетом того, что энтропия имеет размер приведенной теплоты, для бесконечно малой стадии процесса данное утверждение записывают так:
Это означает, что процесс может идти только в том случае, если величина слева — отрицательна или равна нулю.
б) Если теплота выделяется, то ![]() что дает отрицательный вклад в баланс.
что дает отрицательный вклад в баланс.
Аналогично, если энтропия увеличивается, то –T dS тоже меньше 0 (поэтому член T dS взят с обратным знаком). Оба эти изменения способствуют протеканию процесса.
в) Выше говорилось, что процесс может идти за счет и лишь одного стимула, если он «перевешивает» неблагоприятное воздействие второго критерия.
![]() г) Поскольку
г) Поскольку 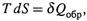 то второе начало термодинамики можно записать и так:
то второе начало термодинамики можно записать и так:
Следовательно, с учетом знака теплота реального процесса — не больше теплоты обратимого процесса, связывающего те же состояния системы.
2. Кроме того, можно объединить первое и второе начала термодинамики в одном выражении.
а) Запишем первое начало (1.7) в дифференциальной форме:
выразим отсюда ![]() и подставим в неравенство второго начала (3.31):
и подставим в неравенство второго начала (3.31):
Это – так называемое основное неравенство термодинамики, объединяющее оба ее начала.
б) Из последнего неравенства, в частности, следует, какую предельную по величине работу может совершить система в процессе:
![]()
(Поскольку работа совершается системой, то  так что слева — положительная величина.)
так что слева — положительная величина.)
3. Нередко утверждают, что существование энтропийного фактора ограничивает возможность системы совершать работу.
а) В общем случае это неверно. Как видно из соотношения (3.35), если dS >0 (энтропия возрастает), то справа оказывается бóльшая величина, т. е. повышается верхний предел работы.
б) Действительно, в процессах жизнедеятельности работа часто совершается за счет только градиента концентраций; происхождение энергии здесь целиком обусловлено энтропийным фактором: внутренняя энергия при выравнивании концентраций разбавленных растворов не меняется.
4. а) В тепловых же машинах, с анализа которых зародилась термодинамика, происходит циклический процесс (как было показано на примере цикла Карно; п.3.2). Причём работа совершается за счёт подводимой извне теплоты (Q1), часть которой (Q2) отдаётся системой холодильнику.
б) Ранее (см. (3.8,а)) было найдено, что
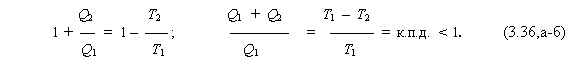 |
Перевернём это равенство и прибавим к каждой его части единицу. Тогда получим:
Здесь учтено, что, в соответствии с (3.6,а-б),
где wпол – результирующая полезная работа, совершённая машиной, а отношение wпол к полученной теплоте (Q1 ) есть коэффициент полезного действия.
в) Неравенство к. п.д. < 1 следует из предположения, что абсолютный нуль температуры недостижим (т. е. Т2 ≠ 0 ).
Действительно, из последнего вытекает, что в тепловой машине даже в обратимом процессе всё получаемое извне тепло трансформироваться в работу не может (что и означает: к. п.д. тепловой машины – меньше единицы).
г) Обычно это утверждение и выражающее его соотношение (3.36) рассматривают как одну из формулировок второго начала термодинамики. Однако суть такого постулата узка и специфична: она справедлива лишь для тепловых машин.
Краткое содержание главы 3
1. В главе дано определение ВТОРОГО НАЧАЛА ТЕРМОДИНАМИКИ: имеются ДВА СТИМУЛА для самопроизвольного изменения термодинамической системы – I. ВЫДЕЛЕНИЕ ТЕПЛОТЫ и II. УВЕЛИЧЕНИЕ ЭНТРОПИИ. Результирующий же критерий, определяющий возможность процесса, есть баланс действия данных стимулов.
2. Фигурирующая здесь ЭНТРОПИЯ – это мера энергетического беспорядка в системе. Но практические её расчёты основаны на соотношении
которое утверждает, что изменение энтропии в любом реальном процессе измеряется приведённой теплотой обратимого процесса, связывающего те же состояния системы.
3. Для доказательства того, что δQобр/T – ФУНКЦИЯ СОСТОЯНИЯ, был рассмотрен цикл Карно.
4. Получен ряд формул расчёта ΔS и S.
I. Нагревание системы:
![]()
II. Абсолютная энтропия:
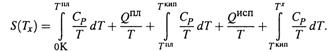
![]() III. Изотермическое расширение газа и изотермическое изменение концентрации раствора:
III. Изотермическое расширение газа и изотермическое изменение концентрации раствора:
5. Рассмотрена также статистическая природа энтропии. Энтропия определяется через ВЕРОЯТНОСТЬ МИКРОСОСТОЯНИЙ (рi) или (при равенстве рi) через ЧИСЛО МИКРОСОСТОЯНИЙ (Ω = 1/p ):
![]()
6. Дана также математическая формулировка второго начала термодинамики:
Это – запись утверждения о том, что термодинамическая возможность процесса определяется балансом двух критериев – теплоты и изменения энтропии.
Глава 4. ТЕРМОДИНАМИЧЕСКИЕ ПОТЕНЦИАЛЫ.
ЭНЕРГИЯ ГИББСА
Применим второе начало термодинамики к различным системам и процессам.
4.1. Изолированные системы
1. В изолированных системах, как известно, исключён обмен энергией с окружающей средой. Поэтому в неравенстве (3.31), выражающем второе начало термодинамики, следует положить
откуда
Таким образом, здесь остается только 1 стимул — увеличение энтропии. Самопроизвольными являются лишь процессы, ведущие к увеличению общей энтропии.
![]() 2. Вселенная в целом, как уже отмечалось в (п. 1.1), — изолированная система. Получается, что все самопроизвольные процессы ведут к увеличению общей энтропии Вселенной. Этот вывод не нов, поскольку данное утверждение постулировалось неравенством (3.3):
2. Вселенная в целом, как уже отмечалось в (п. 1.1), — изолированная система. Получается, что все самопроизвольные процессы ведут к увеличению общей энтропии Вселенной. Этот вывод не нов, поскольку данное утверждение постулировалось неравенством (3.3):
Cправедливости ради заметим, что, видимо, во Вселенной возможны и процессы, дающие обратный результат — концентрацию огромных количеств энергии и массы в небольших объемах пространства.
4.2. Закрытые системы: изотермо-изохорные процессы
1. а) Известно (п. 1.1), что компоненты химической реакции можно рассматривать как закрытую систему. Кроме того, многие реакции (особенно биологические) проходят при постоянной температуре.
Поэтому ситуации, которые предстоит рассмотреть в этом и в следующем пунктах, являются для химика и биолога наиболее важными.
б) Вследствие постоянства температуры, выражение второго начала термодинамики можно записать в данном случае в интегральном виде:
![]()
в) Кроме температуры, во многих процессах остается постоянным какой-либо другой ключевой параметр — объем или давление. В этих случаях критерий (4.2) еще несколько модифицируется.
2. а) Пусть постоянным является объем. Работа против давления тогда не совершается. В отсутствие какой-либо иной работы теплота процесса, в соответствии с (1.11,б), равна изменению внутренней энергии:
![]()
вследствие чего неравенство (4.2) превращается в следующее:
![]()
б) А это позволяет ввести новую функцию состояния — свободную энергию (или просто энергию) Гельмголъца:
в) При постоянной температуре ее изменение равно
![]()
т. е. совпадает с левой частью неравенства (4.3).
Следовательно, второе начало термодинамики, применительно к изотермо-изохорным процессам, можно записать так:
г) Итак, самопроизвольными являются такие процессы, в которых энергия Гельмгольца убывает (не возрастает). Введение особой функции позволяет, как и для изолированных систем, пользоваться лишь одним — в данном случае суммарным — критерием для исследования способности системы к процессам.
Однако этот чисто формальный прием не отменяет того факта, что на самом деле для самопроизвольных процессов по-прежнему остаются те же два стимула; просто через ΔA обозначен их баланс.
4.3. Закрытые системы: изотермо-изобарные процессы
Ещё более важны для нас изотермо-изобарные процессы.
а) Из-за постоянства температуры интегральная форма второго начала термодинамики остаётся справедливой и здесь:
![]()
б) Однако теперь теплота, в соответствии с (2.8), равна изменению энтальпии:
Qp = ΔЕ + P ΔV = ΔН
(при условии, что не совершается никакой иной работы, как против давления).
в) Подстановка в (4.2) приводит к неравенству:
что даёт основание к введению ещё одной функции состояния – пожалуй, наиболее важной в химии – энергии Гиббса (или свободной энергии Гиббса):
G ≡ H – T S . (4.7,a)
Как и в случае энергии Гельмгольца, смысл такого определения состоит в том, что при постоянной температуре изменение введённой функции
совпадает с левой частью соответствующего неравенства – в данном случае, (4.6).
г) В итоге получается единый, но опять-таки суммарный, критерий:
Данное выражение означает, что в изотермо-изобарных условиях самопроизвольными являются такие процессы, в которых функция Гиббса убывает (не возрастает). Но опять-таки, ΔG – лишь баланс всё тех же двух стимулов: выделения теплоты и возрастания энтропии.
4.4. Понятие о термодинамических потенциалах
1. Функция состояния, по изменению которой можно судить о возможности или невозможности процесса, называется термодинамическим потенциалом (по аналогии с электрическим потенциалом).
а) Как нам уже известно, в зависимости от типа системы и процесса роль термодинамического потенциала выполняют разные функции состояния:
- в изолированных системах — энтропия,
- в изотермо-изохорных процессах — энергия Гельмгольца,
- в изотермо-изобарных процессах — энергия Гиббса.
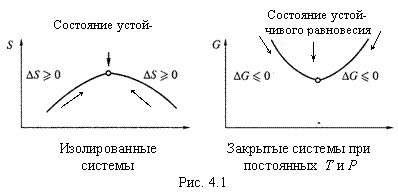 |
б) Можно убедиться также, что в процессах, в которых постоянны энтропия и объем, потенциалом является внутренняя энергия, а в процессах с постоянными энтропией и давлением — энтальпия.
2. В ходе самопроизвольного процесса соответствующий термодинамический потенциал возрастает (если это энтропия) или убывает (любая из остальных вышеперечисленных функций, если она является критерием самопроизвольности). При этом потенциал стремится к некоему экстремальному значению (максимуму в случае энтропии или минимуму в прочих случаях), которому соответствует равновесное состояние системы (рис. 4.1).
Поскольку наиболее важное значение в качестве термодинамического потенциала будет иметь энергия Гиббса, остановимся подробнее на смысле этой величины.
4.5. Смысл и использование энергии Гиббса
Для определения термодинамических «возможностей» изотермо-изобарного процесса в закрытой системе следует опираться на изменение энергии Гиббса (4.7, б):
1. а) По знаку ΔG изотермо-изобарные процессы подразделяются на три группы:
- экзергонические — ΔG < 0, могут идти самопроизвольно;
- обратимые — ΔG = 0, занимают пограничное положение;
- эндергонические — ΔG > 0, сами проходить не могут.
б) Эти термины не надо путать с понятиями экзо - и эндотермичности (п. 2.3), указывающими на знак теплоты процесса.
в) Функция G называется энергией, так как определяет работоспособность соответствующего процесса. А именно, ΔG численно равно максимальной полезной работе, которая могла бы быть совершена в ходе изотермо-изобарного процесса. Это подтверждают следующие выкладки.
2. а) Согласно п. 3.6, в общем случае работа системы в микропроцессе определяется дифференциальным неравенством
![]()
При постоянной температуре его можно переписать для конечных приращений:
![]()
б) Представим левую часть в виде суммы работы расширения и любой другой работы, обозначаемой как полезная:
![]()
В правой же части выразим ΔE через ΔG:
![]()
откуда
![]()
в) Подстановка обоих выражений в неравенство (4.10) дает:
![]()
или
Это и означает, что абсолютное (положительное) значение полезной работы, совершаемой за счет процесса, не больше ΔG (также взятого со знаком плюс).
г) Например, если ΔG0 = –200 кДж/моль, то и работу за счет процесса можно совершить не более 200 кДж/моль.
3. а) Важное замечание: сопряжение экзергонического процесса с какой-либо работой уменьшает (по модулю) результирующее изменение энергии Гиббса; оно становится равным
![]()
так что степень экзергоничности снижается.
б) Пример: анаэробное превращение глюкозы в молочную кислоту:
![]()
В организме человека оно сочетается с совершением полезной работы — образованием молекул аденозинтрифосфата (АТФ) из аденозиндифосфата (АДФ) и фосфат-иона (Фн):
![]()
Поэтому результирующее изменение энергии Гиббса — по модулю меньше, чем в исходном процессе (4.14, а):
![]()
Образующийся АТФ служит источником энергии в различных процессах.
4. При совершении же максимально возможной работы результирующий процесс становится обратимым:
5. Следует также подчеркнуть: поскольку в энергию Гиббса в качестве составной части входит внутренняя энергия:
то абсолютное значение энергии Гиббса различных систем (веществ) неизвестно, вследствие чего оперируют только изменениями этой величины.
4.6. Производные энергии Гиббса по температуре
и давлению
Выше шла речь об энергии Гиббса применительно только к изотермо - изобарным процессам в закрытых системах, где данная функция является термодинамическим потенциалом.
Однако величину ΔG можно рассматривать в любом процессе. От каких параметров и как тогда она зависит?
а) Согласно определению (4.7, а),
![]()
б) Отсюда изменение энергии Гиббса на микростадии произвольного процесса таково:
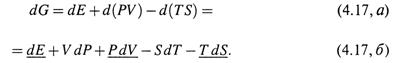
в) Пусть процесс является обратимым (т. е. на каждой микростадии система находится в состоянии равновесия) и пусть возможная работа совершается только против давления. Тогда, в соответствии с (1.7) и (3.4),
![]()
г) С учетом этого в выражении (4.17,б) три подчеркнутые члена сокращаются, давая в сумме ноль. Остается следующее:
![]()
Отсюда вывод: энергия Гиббса равновесного процесса (и равновесной системы) зависит от двух независимых переменных — давления и температуры (чего, в общем, и следовало ожидать).
д) Соответствующие частные производные таковы:
![]()
т. е. температурная зависимость энергии Гиббса определяется энтропией системы, а зависимость от давления — объемом системы.
4.7. Химический потенциал
1. а) Как известно, в химических процессах меняется и количество того или иного вещества. Поэтому энергия Гиббса системы в состоянии равновесия зависит также от количества ее компонентов:
![]()
Это означает, что выражение (4.19) следует расширить, добавляя члены, содержащие дифференциалы ni :
![]()
б) Множители μj при данных дифференциалах называются химическими потенциалами компонентов системы.
Очевидно, каждый такой множитель — это частная производная энергии Гиббса по количеству соответствующего вещества:
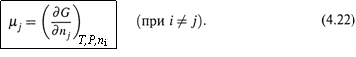 |
2. а) Если условия являются изотермо-изобарными (т. е. когда dP = dT = 0), выражение (4.21,б) сводится к сумме парциальных энергий Гиббса, относящихся к отдельным химическим компонентам системы:
![]()
б) Отсюда вытекает простая интерпретация химического потенциала того или иного компонента. По существу, μj — это та часть энергии Гиббса системы, которая приходится на 1 моль данного вещества, т. е. это просто энергия Гиббса 1 моля вещества.
в) Следовательно, для изотермо-изобарных процессов химический потенциал компонента системы можно отождествлять с его молярной энергией Гиббса, а так как зависимость Gj от числа молей — линейная, то можно просто написать:

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 |
