


Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
в) В то же время для каждого из этих радикалов на основании схемы (20.2) можно составить уравнение «баланса» образования и исчезновения. Сделаем это для радикалов Н∙ :
Отсюда находим стационарную концентрацию данных радикалов:
г) Теперь, используя ту же схему (20.2), запишем уравнение «баланса» для конечного продукта цепной реакции, т. е. HBr:
Как видно, здесь фигурируют те же три члена, что и в уравнении для радикалов Н∙, но из них два последних – с противоположными знаками.
д) I. Подставляя с(Н∙) из (20.11), вынося общий множитель за скобки и приводя выражение в скобках к общему знаменателю, получаем:
II. Наконец, делим числитель и знаменатель на k3∙c(Br2) и, исходя из (20.8,б), учитываем, что
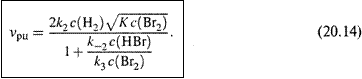 |
е) I. Это дает окончательное выражение:
Именно таким является в данном случае кинетическое уравнение скорости.
II. Не зная же цепного механизма этой реакции и исходя лишь из суммарного
уравнения
Br2 + H2 → 2HBr, (20.1)
мы записали бы совсем иное выражение:
2. Формула (20.14) весьма поучительна и интересна.
а) Так, она показывает, что скорость итоговой реакции зависит от концентраций не только реагентов, но и продукта (HBr).
Это следствие того, что одна из стадий образования данного продукта
является обратимой. То же самое, по существу, мы имели в п. 17.2 для простейшей обратимой реакции первого порядка:
б) Кроме того, порядок реакции по Br2 является дробным. Причем, он меняется по мере расходования этого реагента.
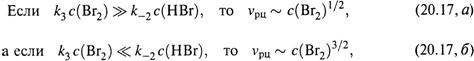 |
т. е. порядок реакции по Br2 возрастает от 1/2 до 3/2. Подобное явление мы
наблюдали и для фотохимических процессов (п. 19.6).
20.3. Общее представление о каталитических реакциях
1. а) Как известно, катализатор — это вещество, изменяющее скорость ре-
акции, но не расходуемое в ходе данной реакции.
б) Катализаторы белковой природы называются ферментами (или энзимами). Практически все реакции в живых организмах идут с достаточной скоростью при такой невысокой температуре, как температура тела, только оттого, что ускоряются ферментами.
2. а) По тому, как именно изменяется скорость реакции в присутствии катализатора, иногда различают два типа катализа – положительный (повышение скорости) и отрицательный (снижение скорости).
б) Однако гораздо чаще термин «катализ» употребляют без подобного уточнения, имея в виду лишь положительный вид катализа. А снижение скорости реакции под действием какого-либо вещества называют ингибированием.
3. Кроме того, катализ подразделяют на гомогенный и гетерогенный.
а) В первом случае катализатор находится в той же фазе, что и реагирующее вещество; обычно это жидкая или газовая фаза.
б) При гетерогенном катализе катализатор образует иную (как правило, твердую) фазу; участники же реакции находятся в газовой или жидкой среде.
Тогда реакция проходит на поверхности раздела фаз. Подобный тип катализа используется в промышленности. Мы же далее будем иметь в виду, в основном, гомогенный катализ.
4. Наконец, иногда каталитическим действием обладает какой-либо из продуктов реакции. Такие процессы называются автокаталитическими. Отличие от цепных реакций состоит лишь в природе катализирующего агента: последний не является свободным радикалом.
5. Между тем, свободные радикалы могут образовываться на промежуточных
стадиях каталитического процесса.
а) В качестве примера приведем разложение пероксида водорода:
I. Если в чистом растворе скорость его близка к нулю, то в присутствии ионов Fe2+ процесс резко ускоряется. Начинает функционировать следующий механизм:
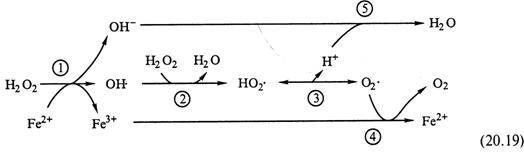 |
Как видно, всего здесь — 5 реакций, суммарное уравнение которых совпадает с уравнением (20.18).
II. Следовательно, ионы Fe2+ выступают в качестве катализаторов. Конкретно, они являются промежуточными донорами электронов, которые необходимы для запуска разложения H2O2.
III. Свободные же радикалы (OH·, HO2· и O2·) образуются лишь на промежуточных стадиях, а не инициируют (как ионы Fe2+) очередной цикл разложения. Поэтому процесс рассматривается как каталитический, а не цепной.
б) Более проста совокупность промежуточных реакций в другом примере.
I. Речь идет о разложении паров ацетальдегида при t ≈ 500°С:
В присутствии паров I2 скорость разложения возрастает примерно в 6000 раз.
II. Механизм катализа включает в данном случае лишь 2 стадии:
III. Здесь катализатор (иод) образует промежуточные соединения с компонентами исходного реагента.
20.4. Особенности катализа
Приведенные примеры позволяют сформулировать общие свойства каталитических процессов.
1. Основное из них состоит в том, что в суммарное уравнение реакции катализатор не входит.
2. Отсюда вытекает ряд принципиальных следствий.
а) В ходе реакции катализатор не расходуется (то, что отмечалось нами в самом определении катализатора).
б) Результирующее изменение энергии Гиббса в обоих вариантах реакции (без катализатора и с катализатором) одно и то же:
в) Если реакция — обратимая, то из предыдущего утверждения следует, что не меняется и константа равновесия (поскольку она непосредственно связана с ![]() ):
):
Иными словами, катализатор не сдвигает положения равновесия в обратимой реакции.
г) Но Kр = kпр /kобр. Поэтому, если в присутствии катализатора Kр остается неизменной, а kпр возрастает, то точно в такое же число раз возрастает и kобр,
т. е. скорости прямой и обратной реакций увеличиваются в одинаковой степени.
д) Два последних утверждения можно объединить в одно: катализатор только ускоряет (за счет увеличения kпр и kобр) достижение того же (что и в его
отсутствие) положения равновесия.
3. Итак, катализатор не входит в суммарное уравнение реакции. Но, как мы
видели из примеров, он непосредственно участвует в тех или иных промежуточных реакциях.
Следовательно, механизм катализа состоит в том, что исходная реакция
(с низкой константой скорости) заменяется на серию других (с более высокими
константами скорости).
20.5. Три принципиальных способа ускорения реакций
Почему же в промежуточных реакциях могут быть более высокие константы скорости? Прежде чем ответить на этот вопрос, для произвольной двусубстратной реакции вида
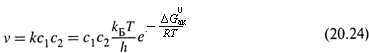 |
запишем уравнение скорости, используя формулу Эйринга (18.40):
Отсюда можно сформулировать три принципиальных способа ускорения реакций
1. Первый из них — увеличение эффективной концентрации реагентов.
а) Данный механизм явно отсутствует в приведенных двух примерах катализа низкомолекулярными веществами (ионами Fe2+ и молекулами I2).
Но он может играть важную роль, когда поверхность катализатора достаточно велика и на ней предварительно связываются молекулы реагентов. Такая ситуация имеет место при гетерогенном катализе, а также при ферментативном катализе, осуществляемом макромолекулами (каковыми являются белки).
в) Так вот, при связывании молекул реагентов с катализатором могут происходить их сильное сближение друг с другом и правильная ориентация друг относительно друга, что и означает повышение эффективной концентрации.
2. Второй принципиальный способ ускорения касается энергии активации.
а) Вспомним её определение (п. 18.2):
т. е. это разница между средней и барьерной (в отношении реакционной активности) энергией вещества.
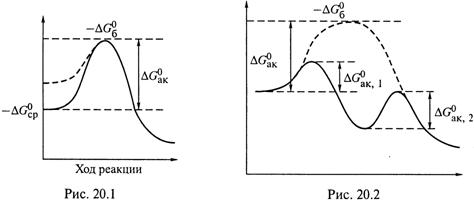
б) Отсюда второй способ ускорения — это уменьшение энергии активации
«снизу», т. е. за счет увеличения средней энергии реагентов (![]() ; рис. 20.1).
; рис. 20.1).
Этот способ тоже возможен лишь в случае катализаторов с достаточно
большой поверхностью (ферменты; гетерогенный катализ).
в) Можно представить, что связывание реагента с катализатором сопровождается созданием напряжения в молекуле реагента. Из-за взаимодействия с химическими группами катализатора, определенная связь в реагенте как бы растягивается» (ослабевает), что означает переход реагента в возбуждённое состояние.
г) Когда же превращение молекулы реагента в продукт завершается, часть высвобождающейся в реакции энергии идёт на восстановление прежней структуры катализатора. Таким образом, повышение энергии реагента катализатором совершается как бы «в долг» – в счёт части энергии последующей реакции.
 |
3. а) И, наконец, третий способ ускорения — уменьшение энергии активации «сверху», т. е. путем снижения энергетического барьера (![]() ; рис. 20.2).
; рис. 20.2).
б) Этого можно достичь путем разбиения исходной реакции на несколько других с меньшими значениями барьерной энергии.
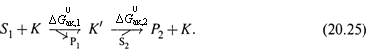 |
Действительно, допустим, что реакция (20.23) заменяется на две другие:
Если соответствующие группы катализатора уже находятся в реакционно-
активном состоянии, то на первой стадии необходимо активирование лишь
реагента S1, а на второй — реагента S2.
в) В исходной же реакции требуется одновременная активация сразу обоих реагентов. Следовательно, энергия активации каждой промежуточной реакции ниже (за счет более низкого барьера) энергии активации исходной реакции. И наиболее высокое из этих небольших значений ![]() определяет константу скорости результирующего превращения.
определяет константу скорости результирующего превращения.
г) Очевидно, именно данный способ реализуется при катализе неорганическими веществами. В случае же ферментов могут использоваться все три способа (хотя, конечно, не обязательно, что все способы — сразу).
4. Продемонстрируем, насколько эффективно влияет снижение ∆G0ак на константу скорости. Пусть под влиянием катализатора ∆G0ак уменьшается от 109 кДж/моль до 74,5 кДж/моль.
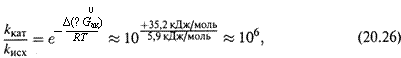 |
Тогда, согласно формуле Эйринга (18.44), при t = 37°С
т. е. скорость реакции увеличивается в миллион раз.
20.6. Уравнение Михаэлиса-Ментен
Теперь обратимся к кинетике каталитических реакций. Для определенности, будем иметь в виду ферментативные реакции, хотя многое из нижеследующего справедливо и для прочих каталитических процессов.
1. Как и для цепных реакций, уравнение скорости ферментативного процесса зависит от его конкретного механизма. Рассмотрим простейший случай: односубстратныв необратимые реакции вида
протекающие в две стадии:
На первой (обратимой) стадии образуется фермент-субстратный комплекс ES, а на второй он необратимо распадается на фермент и продукт реакции.
2. Вывод уравнения скорости производится по тому же принципу, что и для
цепных реакций (п. 20.2).
а) Так, тоже исходят из условия стационарности. В данном случае оно означает постоянство концентрации комплекса ES:
б) Заметим, что концентрация свободного фермента равна
где cE,0 — общая концентрация фермента — и в свободном состоянии, и в составе комплекса ES.
в) Подставляя cE в (20.29), можно найти стационарную концентрацию комплекса:
Здесь введена константа Михаэлиса:
г) Скорость же образования конечного продукта, а значит и скорость реакции в целом, очевидно, такова:
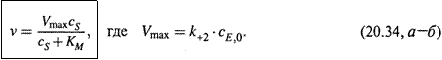 |
д) Подставляя cES, получаем искомое выражение:
Величина Vmax — это максимальная скорость реакции; формула же (20.34, а) — уравнение Михаэлиса—Ментен, являющееся основным в ферментативной кинетике.
3. Проанализируем это уравнение.
а) При малых концентрациях субстрата S (когда cS « KM) имеем:
т. е. фермент работает в линейном режиме, а реакция имеет первый порядок.
б) Если же, напротив, концентрация субстрата очень велика, получаем режим насыщения:
Здесь скорость перестает зависеть от концентрации субстрата, не поднимаясь выше Vmax, т. е. реакция приобретает нулевой порядок.
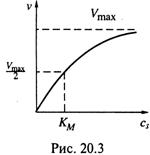
в) Отсюда — та гипербола, которой описывается зависимость v от ![]() для ферментативной реакции (рис. 20.3). По мере же расходования субстрата порядок реакции изменяется от нулевого до первого.
для ферментативной реакции (рис. 20.3). По мере же расходования субстрата порядок реакции изменяется от нулевого до первого.
20.7. Временные характеристики ферментативной реакции
Явление насыщения — характерное свойство не только ферментативных,
но и вообще всех каталитических реакций.
1. Чтобы лучше понять его природу, полезно ввести в рассмотрение временные характеристики: Т — период деятельности фермента, подразделяемый на 2 части:
T = tχ + tр. (20.36)
а) При этом tχ — холостое время, или время «простоя» молекулы фермента,
т. е. это среднее время от высвобождения с фермента молекулы продукта до
результативного связывания с ферментом очередной молекулы субстрата.
б) А tр — рабочее время, или время собственно ферментативного акта. Иначе говоря, это среднее время от результативного связывания субстрата до высвобождения продукта.
2. а) В ненасыщающем режиме отличны от нуля и tχ, и tр.
б) Но холостое время, очевидно, обратно пропорционально концентрации субстрата:
Действительно, чем выше cS, тем быстрей происходит занятие освободившегося фермента очередной молекулой субстрата. Поэтому
в) Таким образом, в состоянии насыщения фермент (или иной катализатор)
функционирует с максимально возможной для него скоростью, определяемой
только рабочим временем (которое от cS не зависит). Поэтому и пропадает
зависимость от cS.
3. Часто используют число оборотов фермента Aоб. Это количество молекул
субстрата, перерабатываемых молекулой фермента за 1 с. Легко убедиться в
следующих соотношениях:
20.8. Обсуждение уравнения Михаэлиса-Ментен
Сделаем еще несколько замечаний по поводу основного уравнения фер-
ментативной кинетики.
1. Параметры уравнения — Vmax и KM.
а) Смысл величины Vmax мы уже отмечали.
Это максимальная скорость, достигаемая при насыщающих концентрациях
субстрата. Согласно (20.34,б) и (20.39,б),
т. е. Vmax зависит от концентрации фермента и максимального числа оборотов отдельных молекул фермента.
б) Смысл KM таков.
I. Положим cS = KM; тогда из уравнения (20.34, а) вытекает:
Следовательно, KM — такая концентрация субстрата, при которой достигается скорость реакции, равная половине максимальной.
II. Несколько упрощая, можно сказать, что KM отражает сродство фермента к
субстрату: чем это сродство выше, тем KM меньше, т. е. тем круче идет кривая
зависимости υ от cS на рис. 20.3.
в) Итак, KM характеризует сродство фермента к субстрату, а Vmax — активность фермента, уже связавшего субстрат.
2. Определение Vmax и KM.
а) Эти параметры обычно определяют экспериментальным путем, для чего измеряют скорость реакции при разных концентрациях субстрата.
б) Но непосредственно по графику υ(cS) трудно заключить, чему равна Vmax: не всегда ясно, к какому пределу стремится кривая. Поэтому преобразовывают уравнение Михаэлиса—Ментен к линейной форме.
в) Наиболее часто используется метод Лануйивера—Берка (или метод обратных величин):
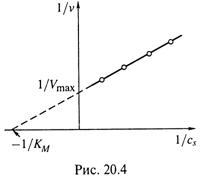
Здесь получается линейная зависимость 1/υ от 1/cS.
г) Итак, по экспериментальным данным строят график для обратных величин (рис. 20.4). И, продолжая его, находят точки пересечения с осями координат. Как видно из рис. 20.4, эти точки отсекают на оси абсцисс величину –1/KM, а на оси ординат – 1/Vmax.
3. Решение уравнения Михаэлиса–Ментен.
а) Если учесть, что
то уравнение Михаэлиса–Ментен — это дифференциальное уравнение относительно cS:
б) Здесь можно разделить переменные и проинтегрировать:
откуда
 Два первых члена правой части — это постоянная интегрирования, опре-
Два первых члена правой части — это постоянная интегрирования, опре-
деляемая из начального условия (при t = 0 cS = 0).
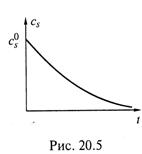
в) Но полученное алгебраическое уравнение является трансцендентным, и в явном виде выразить из него ![]() нельзя. Тем не менее, видно, что при росте t cS уменьшается, что примерно отражается графиком на рис. 20.5. Начальная часть графика близка к линейной зависимости {нулевой порядок реакции), а
нельзя. Тем не менее, видно, что при росте t cS уменьшается, что примерно отражается графиком на рис. 20.5. Начальная часть графика близка к линейной зависимости {нулевой порядок реакции), а
конечная — к экспоненциальной {первый порядок).
Краткое содержание главы 20
В главе рассмотрены цепные и каталитические реакции.
1. а) Особенностью ЦЕПНЫХ реакций является то, что среди их продуктов образуются СВОБОДНЫЕ РАДИКАЛЫ, которые вовлекают в процесс новые молекулы реагентов.
В самом процессе различают три стадии – зарождение, продолжение и обрыв цепи. Стадия продолжения состоит из повторяющихся циклов реакций (т. н. звеньев), инициирующих друг друга.
б) Были приведены примеры цепных реакций, в т. ч.:
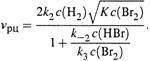 |
в) Кинетические уравнения скорости зависят от механизма цепного процесса. В частности, для первой из приведенных реакций уравнение таково:
По мере расходования брома порядок реакции по нему возрастает от 1/2 до 3/2.
2. а) Что касается КАТАЛИТИЧЕСКИХ процессов, то даны краткие представления о ПОЛОЖИТЕЛЬНОМ и отрицательном, ГОМОГЕННОМ и ГЕТЕРОГЕННОМ катализе, АВТОКАТАЛИТИЧЕСКИХ процессах, ФЕРМЕНТАХ – биологических катализаторах белковой природы.
б) Принципиальная особенность катализа состоит в том, что катализатор не входит в суммарное уравнение реакции и увеличивает в равной степени скорость как прямой, так и обратной реакции.
в) Возможные способы действия катализаторов таковы:
- увеличение эффективной концентрации реагентов,
- повышение энергии реагентов (создание напряжения в их молекулах),
- понижение энергетического барьера (путём разбиения исходной реакции на промежуточные стадии с меньшими барьерами).
г) Для ферментативных реакций, протекающих по простейшему механизму:
скорость описывается УРАВНЕНИЕМ МИХАЭЛИСА-МЕНТЕН:
д) При этом КОНСТАНТА МИХАЭЛИСА, КМ , численно равна концентрации субстрата, при которой скорость реакции составляет половину максимальной (0,5 Vmax).
Значения параметров КМ и Vmax можно определить по графику зависимости 1/v от 1/cS (метод Лануйивера-Берка).
е) Интегрирование же уравнения Михаэлиса-Ментен приводит к трансцендентному алгебраическому уравнению относительно концентрации субстрата:
cS + KM∙ ln cS = coS + KM∙ ln coS – Vmax .
Глава 21. КИНЕТИКА ГЕТЕРОГЕННЫХ ПРОЦЕССОВ
21.1. Введение
До сих пор речь шла о кинетике лишь гомогенных процессов.
1. Теперь же обратимся к гетерогенным процессам, где реагирующие вещества находятся в разных фазах. Следовательно, в таких процессах реакция происходит на границах раздела фаз.
 2. Конкретным примером являются электродные процессы (главы 14–15) —
2. Конкретным примером являются электродные процессы (главы 14–15) —
электролиз и генерация ЭДС в гальванических элементах.
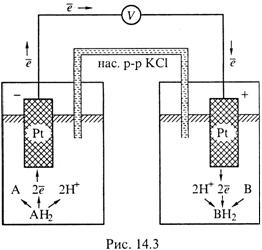
В частности, возьмем элемент из двух редокс-электродов (рис.14.3).
В левом полуэлементе взаимодействуют растворенное вещество AH2 и платина в составе твердого электрода: эти вещества обмениваются электронами.
Очевидно, сама полуреакция происходит на поверхности электрода.
То же можно сказать о правом полуэлементе.
3. Реакции на границе раздела фаз имеют определенные особенности кинетики. Эти особенности мы обсудим вначале с общих позиций (т. е. применительно к любым гетерогенным процессам), а в следующей главе — в отношении конкретных электрохимических процессов.
а) В гетерогенной реакции различают обычно не менее трех стадий:
I. перенос реагирующих веществ к поверхности раздела фаз,
II. собственно реакцию,
III. отвод продуктов реакции от поверхности.
б) Причём в основе первой и третьей стадий лежат не химические, а физико-химические процессы. Как правило, это диффузия и движение частиц в электрическом поле.
4. В частности, для приведенного выше примера стадии левой полуреакции отражаются схемой:
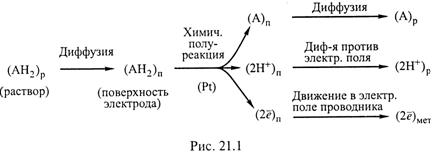 |
5. а) Результирующая же скорость гетерогенной реакции определяется, очевидно, самой медленной стадией. Чаще всего такой стадией является именно диффузия.
б) Следовательно, диффузия играет очень важную роль в кинетике гетерогенных процессов. В связи с этим, остановимся на ней подробнее.
21.2. Общие сведения о диффузии
1. а) Диффузия — это самопроизвольное перемещение частиц {молекул) из области с более высокой в область с более низкой концентрацией. В основе ее — хаотичное тепловое движение данных частиц.
б) В п. 4.8 мы давали практически такое же определение для осмотических
процессов. И находили, что при переходе п молей вещества от концентрации c1
к меньшей концентрации c2 уменьшение энергии Гиббса системы равно
в) Отсюда следуют два обстоятельства.
I. Во-первых, диффузия — это те же осмотические процессы, но рассматриваемые на уровне частиц и молекул.
II. Во-вторых, в результате диффузии, как и в осмотических процессах, теплота не поглощается и не выделяется, а все изменение ∆G обусловлено изменением только энтропии системы.
г) Причем, видимо, в ходе диффузии энтропия возрастает, т. к. частицы переходят к менее упорядоченному расположению. Таким образом, диффузия — одно из проявлений второго начала термодинамики.
2. а) Важнейшей характеристикой является поток диффузии в каком-либо на-
правлении, jдиф. Это количество вещества (в молях), диффундирующего в единицу времени через единичное поперечное сечение, перпендикулярное данному направлению:
3. Для диффузии характерны следующие три закона.
I. Первый закон Фика (в одномерном варианте):
а) Суть этого закона в том, что поток диффузии в определенном направлении
пропорционален градиенту концентрации вещества в данном направлении.
Иными словами, чем больше разница концентраций между точками пространства, тем сильней диффузия.
б) Знак минус в формуле (21.2, а) обусловлен тем, что диффузия происходит в сторону снижения градиента концентрации (дс/дx < 0), тогда как величина jдиф должна быть положительной.
в) Что касается D, то это — коэффициент диффузии.
 |
II. Второе соотношение — уравнение непрерывности:
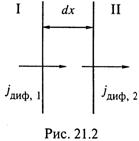
а) Чтобы понять его, выделим на направлении диффузии небольшой интервал dx, ограниченный сечениями I и II (рис. 21.2).
Ввиду малости интервала можно полагать, что во
всех его точках концентрация одинакова и равна с.
б) Так вот, уравнение (21.3) констатирует довольно очевидную вещь.
Концентрация вещества в интервале dx изменяется со временем (дc/дt ≠ 0) в том случае, если на границах этого интервала различен диффузионный поток (д jдиф /дx ≠ 0).
в) Действительно, чтобы, например, концентрация убывала со временем, необходимо, чтобы приток вещества в интервал был меньше оттока вещества из интервала:
г) Таким образом, положительный градиент потока диффузии (∂ jдиф/∂х > 0) вызывает снижение концентрации в точках интервала (∂с/∂t < 0), а отрицательный градиент потока – возрастание концентрации.
III. Наконец, третье соотношение — второй закон Фика. Он следует из первых двух законов, если коэффициент диффузии одинаков во всех точках пространства:
откуда
 |
б) Итак, скорость изменения со временем концентрации вещества в некоторой точке пространства пропорциональна второй производной концентрации вещества по направлению диффузии.
4. Завершая краткое изложение сведений о диффузии, отметим также свойст-
ва стационарной диффузии.
а) Диффузия называется стационарной, если концентрация вещества в точках пространства, несмотря на диффузию, не меняется:

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 |
