


Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
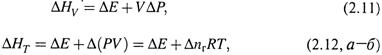 |
4. а) Для изохорного и изотермического процессов изменение энтальпии таково:
где Δnг — изменение количества газообразных веществ.
б) Если все участники реакции — в жидком или твердом состоянии, то величинами ΔР и Δ (PV) можно пренебречь, и тогда
Как видно, для изохорного процесса изменение энтальпии вновь почти совпадает с теплотой.
5. Существует еще одно толкование энтальпии.
Поскольку PV — потенциальная энергия не только газообразных, но и жидких веществ, то Н = Е + PV — это сумма внутренней и потенциальной энергии системы.
Однако важней всего связь ΔН с теплотой.
2.3. Расчёт теплот реакций
Как же рассчитывать теплоты реакций, используя представление об энтальпии?
1. Поскольку в определение H входит Е, то абсолютное значение энтальпии системы (или какого-то вещества) неизвестно.
Поэтому энтальпию вещества характеризуют стандартными энтальпиями образования и сгорания.
а) ΔНообр (стандартная энтальпия образования) — это теплота реакции образования вещества из простых веществ (или элементов) при стандартных условиях:
простые вещества → X . (2.14, а)
При этом для самих простых веществ ΔНообр считается равной нулю.
б) А ΔНосг (стандартная энтальпия сгорания) — теплота реакции сгорания вещества до оксидов (с максимальной степенью окисления) при стандартных условиях:
Х + О2 → оксиды (с макс. СО). (2.14,б)
Здесь для указанных оксидов принимается, что ΔНосг = 0.
в) Величины ΔНосг определены для очень многих веществ экспериментальным способом — путем сжигания вещества в калориметрической «бомбе» и измерения выделяющейся теплоты.
Зная ΔНосг, можно вычислить и ΔНообр.
Эти значения (ΔНосг и ΔНообр) содержатся во многих справочных таблицах.
2. а) Для получения формул, показывающих, как пользоваться этими величинами, обратимся к простейшей реакции:
А → В (2.15)
ΔНрц
Здесь ΔНрц — теплота реакции.
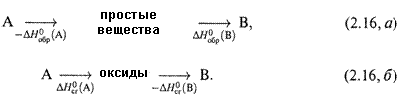 |
б) Теплота результирующего превращения не изменится (по закону Гесса), если его провести одним из двух таких способов:
в) Отсюда получаем:
г) Обобщение этих выражений для произвольных реакций приводит к формулам:
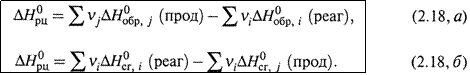 |
Здесь νi и νj — стехиометрические коэффициенты перед соответствующими веществами в уравнении реакции.
д) Таким образом, для расчета теплоты реакции по энтальпиям образования следует из суммы энтальпий продуктов вычесть сумму энтальпий реагентов.
А при использовании энтальпий сгорания — наоборот, из суммы энтальпий реагентов вычесть сумму энтальпий продуктов.
3. Вот пример расчета по этим формулам. Спиртовое брожение глюкозы у дрожжей:
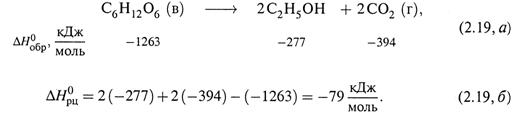 |
4. а) По знаку теплоты (т. е. ΔНорц) реакции подразделяются на
- экзотермические, где ΔНорц < 0 , т. е. теплота выделяется, и
- эндотермические, в которых ΔНорц > 0 — теплота поглощается.
б) Таким образом, приведенный выше процесс брожения глюкозы является экзотермическим.
2.4. Теплоёмкость веществ
Наряду с энтальпиями образования и сгорания непосредственное отношение к теплоте имеет теплоемкость.
1. Дадим определение: молярная теплоемкость вещества – это количество теплоты, требующееся для увеличения температуры 1 моля вещества на 1 градус:
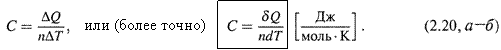 |
(Для обозначения бесконечно или малой теплоты в произвольном процессе здесь и далее используется знак δ, а не d, поскольку теплота может не являться функцией состояния, и тогда величина δQ зависит от способа осуществления данной микростадии процесса).
2. а) Известны два частных вида теплоемкости — CV и CP :
- в первом случае теплоемкость определяется при постоянном объеме,
- во втором — при постоянном давлении.
б) Учтем, что, согласно (1.11,б) и (2.9),
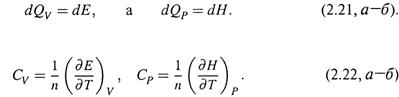
Отсюда
Как видим, CV — это производная внутренней энергии по температуре, отнесенная к 1 молю вещества, а CP — производная энтальпии.
3. Можно также установить связь этих двух величин между собой.
а) При P = const для идеальных газов
б) Подставляя последнее выражение в (2.22,б), получаем:
Таким образом, эти две теплоёмкости различаются всего на величину R ≈ 8,31 Дж/(моль∙К). Причём, это справедливо лишь для идеальных газов; в случае же жидких и твёрдых веществ СР = СV .
4. а) Возвращаясь к идеальному газу, напомним:
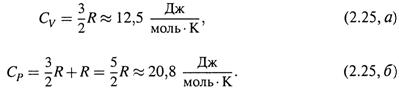 |
б) Поэтому для него, согласно (2.22, а) и (2.24,6),
в) Для сравнения приведем теплоемкость воды в пределах 10°—20° С:
Как видно, она гораздо больше. Так что, в самом деле, оценки (2.25,а-б) справедливы только для идеального газа.
2.5. Зависимость теплоты реакции от температуры
В ряде случаев (хотя не всегда) теплота реакции (величина ΔНорц) зависит от температуры.
1. Наличие этой зависимости и сам ее характер определяется разностью теплоемкостей продуктов и реагентов, то есть (при постоянном давлении) величиной
где ni и nj — по-прежнему стехиометрические коэффициенты реакции.
а) Действительно, пусть данная разность — положительная, т. е. образуются более теплоемкие вещества. Тогда при нагревании системы все большая часть теплоты должна как бы оставаться в продуктах реакции.
Следовательно, выделение теплоты в реакции уменьшается.
А это значит, что величина ΔН (с учетом знака) возрастает: например, с –200 кДж/моль до –100 кДж/моль.
б) Аналогично, если ΔСР < 0, то образуются менее теплоемкие вещества: при нагревании они поглощают меньше теплоты, чем реагенты, отчего больше теплоты выделяется в ходе реакции, а величина ΔНорц уменьшается.
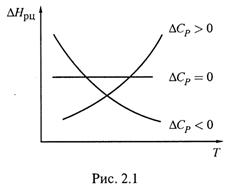 Соответствующие зависимости показаны на рис. 2.1.
Соответствующие зависимости показаны на рис. 2.1.
2. Теперь установим конкретный характер этой зависимости. Вновь обратимся к простейшей реакции А → В..
а) В расчете на 1 моль реагента или продукта находим, с учетом (2.22, б):
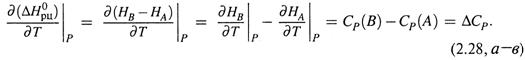 |
б) Тем самым получаем уравнения Кирхгофа:
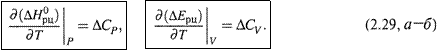 |
Второе из них выводится аналогично первому.
в) Как видим, и для изобарного, и для изохорного процесса производная теплоты по температуре равна ΔС (соответственно, ΔСР или ΔСV). Характерно, что совпадают и знаки этих величин.
г) Поэтому, если ΔСР < 0 (т. е. продукты – менее теплоемкие), то теплота реакции при нагревании снижается.
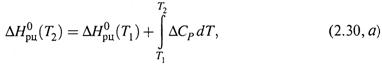 |
3. а) Для практических целей уравнение Кирхгофа используют в интегральной форме:
где ΔНорц — энтальпия реакции при температуре T0 .
б) В принципе, теплоемкости веществ зависят от температуры, отчего ΔСР — тоже какая-то функция температуры. Это затрудняет точное интегрирование в формуле (2.30, а).
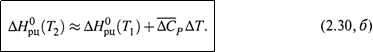 |
Но часто ограничиваются приближенным расчетом, где используют средние значения теплоемкостей веществ на рассматриваемом интервале температур (причем, средние арифметические значения, что и обуславливает приближенность расчета):
в) Итак, зная теплоту реакции при T1, можно оценить теплоту реакции при T2 .
4. Но обычно изменение ΔНорц при нагревании не очень велико.
а) Так, допустим, что
Тогда подстановка в формулу (2.30,б) дает:
Следовательно, теплота реакции на интервале в 50 К изменяется всего на 1 % (cо 100 кДж/моль до 101).
б) Если теплоемкости реагентов и продуктов еще ближе по величине, то и изменение энтальпии будет еще меньше. Поэтому очень часто считают, что ΔНорц практически не зависит от температуры.
2.6. Теплоты физико-химических процессов
Не только химические, но и физико-химические процессы могут сопровождаться выделением или поглощением теплоты.
В связи с этим, рассмотрим три типичных физико-химических процесса.
1. Растворение веществ.
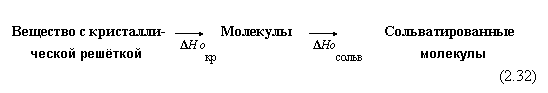 |
а) Если это твердое вещество, то процесс можно представить в две стадии:
На первой стадии разрушается кристаллическая решетка, на второй происходит сольватация (например, гидратация) молекул вещества. Обычно первая стадия — эндотермическая, вторая — экзотермическая:
Результирующая же теплота растворения равна сумме теплот двух стадий:
и может иметь как положительный, так и отрицательный знак.
б) А вот ситуации, когда присутствует только одна из двух названных стадий.
I. При растворении газов, очевидно, нет первой стадии — разрушения
кристаллической решетки. Остается экзотермическая сольватация. Поэтому
растворение газов, как правило, экзотермично.
II. При растворении кристаллогидратов отсутствует стадия сольватации.
Остается лишь эндотермическое разрушение кристаллической решетки. Процесс идет с поглощением теплоты.
в) В качестве примера приведем растворение сульфата меди — в первом случае кристаллогидрата, а во втором — безводной соли:
 |
2. Реакция нейтрализации.
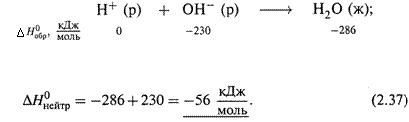 |
а) Сильные кислоты и основания в растворе диссоциированы полностью, и поэтому теплота нейтрализации (независимо от их природы) одна и та же.
Рассчитаем ее:
б) Если же в подобной реакции участвует слабый электролит (у которого диссоциирована лишь небольшая часть молекул), то процесс можно записать в две стадии:
Первая стадия — это ионизация слабого электролита. Она обычно является эндотермической, т. е. для полной ионизации всех молекул слабого электролита требуется передать веществу теплоту. И чем слабее кислота, тем больше надо теплоты.
Итак, в реакции нейтрализации, идущей с участием слабого электролита, результирующее выделение энергии оказывается меньше, чем в случае сильного электролита.
в) Примеры.
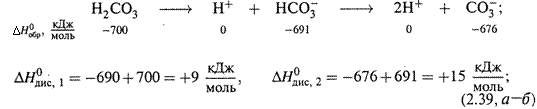 |
I. Диссоциация угольной кислоты:
II. Реакция нейтрализации угольной кислоты щелочью:
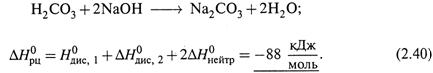 |
3. Фазовые превращения
а) Теплота фазового перехода равна разности ΔНообр вещества в одном и в другом состояниях.
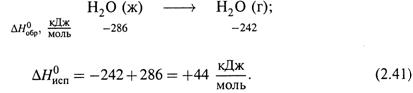 |
б) Например, для испарения воды (при 25 °С) имеем:
Таким образом, парообразование – это эндотермический процесс.
в) Как меняется теплота испарения при повышении температуры? Для ответа на этот вопрос будем считать, что испарение – это аналог химической реакции, и применим уравнение Кирхгофа.
В п. 2.4 мы видели, что теплоёмкость воды («реагента») значительно больше, чем теплоёмкость идеального газа. А пар («продукт реакции») отчасти подобен такому газу. Поэтому
Следовательно, при нагревании теплота испарения снижается.
И действительно – при 100°С
ΔHисп (Н2О) = + 41 кДж/моль < ΔH°исп (Н2О) . (2.43)
Краткое содержание главы 2
1. Для условий, в которых обычно проходят химические реакции, теплота реакции есть функция состояния. Это констатирует ЗАКОН ГЕССА: теплота реакций не зависит от способа их осуществления.
2. Более общим понятием является ЭНТАЛЬПИЯ – такая функция состояния, изменение которой в изобарном процессе равно теплоте:
3. Используя энтальпии (теплоты) образования или сгорания веществ, рассчитывают ТЕПЛОТЫ РЕАКЦИЙ:
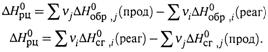 |
4. В главе также введены МОЛЯРНАЯ ТЕПЛОЁМКОСТЬ и два её частных вида:
5. Через эти величины выражается зависимость теплоты реакции от температуры (УРАВНЕНИЯ КИРХГОФА):
6. И, наконец, были рассмотрены теплоты ряда физико-химических процессов – растворения веществ, фазовых превращений и диссоциации слабых электролитов.
Глава 3. ВТОРОЕ НАЧАЛО ТЕРМОДИНАМИКИ
До сих пор мы исходили лишь из первого начала термодинамики. Теперь же перейдем к рассмотрению второго начала и связанных с ним термодинамических функций.
3.1. Общий смысл второго начала
1. Второе начало дает критерии того, какие процессы могут проходить самопроизвольно, а какие — нет.
а) Опыт показывает, что многие процессы идут с выделением теплоты (ΔH < 0).
б) Но среди самопроизвольных встречаются также процессы с почти нулевым тепловым эффектом (ΔH ≈ 0) и даже эндотермические процессы (ΔH > 0). Примером является растворение аммония нитрата в воде:
В этом самопроизвольном процессе происходит заметное поглощение теплоты, так что жидкость в сосуде сильно охлаждается.
в) Следовательно, тепловыделение не является решающим критерием того, может процесс протекать самопроизвольно или нет.
2. Второе начало термодинамики вводит в рассмотрение еще один критерий — энтропию, S, которая служит мерой энергетического беспорядка в системе. Чтобы понять, что это такое, рассмотрим два примера.
а) Распределение молекул вещества по объему.
Допустим, что все молекулы вещества сосредоточены в небольшой части V1 объема Vo (рис. 3.1, а). Это означает наличие в системе высокой упорядоченности, причем упорядоченности энергетической, поскольку молекулы являются носителями внутренней энергии.
Из опыта известно, что такой порядок неустойчив: частицы (если они ничем не зафиксированы) стремятся занять весь объем Vo, т. е. расположиться в нем самым произвольным образом (рис. 3.1,б).
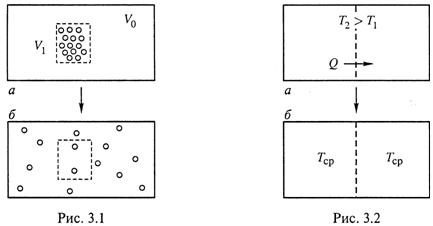 |
В результате в системе повышается степень энергетического беспорядка.
б) Выравнивание температур между частями системы.
Пусть вначале одна часть тела (системы) горячей, чем другая часть. Это тоже означает наличие упорядоченности: частицы с большей энергией находятся в одной части пространства, а частицы с меньшей энергией — в другой.
Хорошо известно, что в такой системе температура частей со временем самопроизвольно выравнивается, т. е. вновь достигается состояние с большим энергетическим беспорядком.
3. а) Напомним, что мера такого беспорядка называется энтропией. Следовательно, в обоих рассмотренных самопроизвольных процессах энтропия системы возрастает:
б) А могут ли происходить процессы, в которых энтропия не увеличивается, а понижается? — Да. Пример — реакции вида
Здесь между частицами устанавливается связь, и поэтому они не могут располагаться в пространстве так же произвольно, как прежде. Следовательно, степень энергетического беспорядка уменьшается. И таких самопроизвольных реакций известно очень много.
4. Так каков же критерий самопроизвольности — теплота или энтропия? Оказывается, в общем случае и то, и другое.
а) Чтобы отразить это, можно сформулировать второе начало термодинамики следующим образом.
Имеются два стимула для самопроизвольного изменения термодинамической системы:
I) снижение суммарной энергии системы (выделение теплоты) и
II) более равномерное распределение энергии по компонентам и пространству системы (увеличение энтропии).
Результирующий же критерий, определяющий возможность процесса, есть баланс действия этих стимулов.
б) В учебниках в качестве второго начала обычно приводится множество других формулировок, например:
«Теплота не может самопроизвольно передаваться от более холодного тела к более горячему»,
«Коэффициент полезного действия паровой машины всегда меньше единицы» и т. д.
Но они, в основном, важны для физических процессов. А для химических процессов, с моей точки зрения, суть дела лучше всего отражается утверждением о двух стимулах.
5. Заметим: из этого утверждения, в частности, следует, что процессы могут идти и под влиянием только одного стимула, если второй отсутствует или даже оказывает противодействующее, но более слабое, влияние. Поэтому возможны
- самопроизвольные процессы, в которых поглощается теплота (если рост энергетического «беспорядка» является более весомым), и
- процессы, в которых понижается энтропия системы (если это сопровождается более сильным выделением теплоты).
6. а) Сделаем еще одно важное замечание. Выделение системой теплоты приводит к увеличению энтропии окружающей среды (из-за ускорения теплового движения частиц среды «беспорядок» их расположения возрастает), поэтому в балансе двух стимулов первый стимул (выделение теплоты) можно заменить на другой — возрастание энтропии внешней среды.
б) Тогда второе начало термодинамики можно сформулировать следующим образом.
Самопроизвольными являются такие процессы, которые приводят к увеличению общей энтропии окружающей среды и системы:
в) Тем не менее при анализе конкретной системы рассматривать изменения в окружающей среде не всегда возможно, потому-то удобней исходить из первоначальной формулировки второго начала.
7. Важно иметь в виду и то, что сделанное утверждение — это условие необходимости, но не достаточности: наличие термодинамических стимулов означает, что процесс может протекать, но не означает, что он действительно будет идти. Последнее зависит уже от кинетических характеристик.
3.2. Связь энтропии с теплотой обратимого процесса
Как конкретно определить изменение энтропии ΔS?
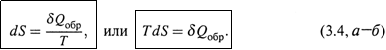 |
1. а) Расчеты основаны на следующем ключевом положении: изменение энтропии измеряется приведенной теплотой обратимого процесса, связывающего соответствующие состояния системы:
Заметим: речь идет о том, что энтропия именно измеряется величиной δQобр/T, но вовсе не тождественна ей по смыслу.
б) Соотношения (3.4) основываются на следующих трех фактах.
I. Приведенная теплота обратимого микропроцесса δQобр/T, в отличие от просто теплоты δQ или даже теплоты обратимого микропроцесса δQобр, является функцией состояния и не зависит от способа перехода из одного состояния в другое.
II. Функция, измеряемая данной величиной, в необратимых процессах в изолированных системах всегда возрастает, а значит, является критерием самопроизвольности процессов.
III. Одновременно эта функция непосредственно связана с числом микроостояний, возможных в системе, т. е. характеризует меру ее энергетического беспорядка.
в) Эти три факта соответствуют приведенным выше представлениям об энтропии — тому, что это функция состояния, причем, такая, которая является критерием самопроизвольных процессов в изолированных системах и отражает степень энергетического беспорядка системы. Отсюда и следует, что величиной δQобр/T измеряется изменение именно энтропии.
2. Но как убедиться в трёх перечисленных свойствах величины δQобр/T?
а) Доказательство второго и особенно третьего свойства достаточно сложно; эти утверждения читателю придётся принять на веру.
б) А вот первое утверждение (о том, что величина δQобр/T – функция состояния) мы рассмотрим более внимательно.
Его доказательство строится на том, что в циклическом процессе результирующее изменение любой функции состояния, очевидно, должно равняться нулю.
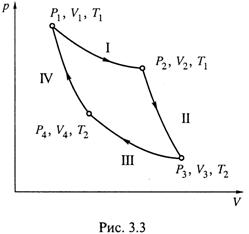
Поэтому обращаются к простейшей газовой системе и рассматривают в ней определённый циклический процесс – цикл Карно. Интеграл функции δQобр/T, действительно, оказывается равным нулю:
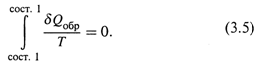
Убедимся в этом.
3. а) Цикл Карно (рис. 3.3) включает 4 обратимые стадии:
I) изотермическое расширение газа от V1 до V2 при температуре T1; поглощается теплота Q1;
II) адиабатическое расширение газа от V2 до V3 (при этом Q = 0, а температура снижается от Т1 до Т2 );
III) изотермическое сжатие от V3 до V4 при температуре Т2; отдается теплота Q2;
IV) адиабатическое сжатие от V4 до V1; вновь Q = 0, а температура увеличивается до T1.
Напомним: адиабатическими называются такие процессы, в которых исключен теплообмен системы с окружающей средой (1.12).
б) Для изотермических стадий цикла воспользуемся формулами (1.8) и (1.6):
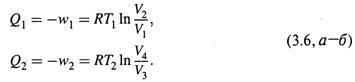 |
в) А исходя из свойств адиабатических процессов, можно доказать, что
г) Учитывая это обстоятельство, поделим почленно соотношения (3.6,а-б) друг на друга:
д)Поскольку для адиабатических стадий Q = 0, получается, что результирующее изменение функции δQобр/T в цикле равно нулю. Что утверждалось соотношением (3.5) и откуда следует, что δQобр/T — функция состояния.
3.3. Дополнительные замечания
Итак, изменение энтропии измеряется приведенной теплотой обратимого процесса (3.4,а):
1. а) Следует обратить внимание на знаки в этом соотношении: если в указанном процессе теплота поглощается системой (δQобр > 0), то dS > 0 — энергетический беспорядок в системе возрастает; если же система отдает теплоту (δQобр < 0), то dS < 0 — беспорядок уменьшается.
2. а) А температурный множитель показывает: если система обратимо получает (или отдает) теплоту при меньшей температуре, на энтропию это влияет сильней.
б) Действительно, пусть при T1 частицы системы имеют энергию по 200 Дж и в ходе процесса отдают в виде теплоты по 20 Дж. Их движения становятся более упорядоченными (dS < 0) всего лишь на 10 %.
Если же это происходит при такой низкой температуре T2, где частицы имеют по 40 Дж энергии, то потеря по 20 Дж скажется на энергетической упорядоченности гораздо сильней.
3.4. Расчет ΔS в некоторых процессах
а) На основании изложенного выше конкретные расчеты изменения энтропии в процессах проводятся по формуле:
 |
б) А для расчета абсолютного значения энтропии (S0, а не ΔS) используют третье начало термодинамики: при абсолютном нуле температуры энтропия любой системы равна нулю:
![]()
т. е. при данной температуре как бы достигается идеальный энергетический порядок. Это положение не доказывается, а принимается в качестве постулата, хотя опирается на определенные экспериментальные факты.
Рассмотрим вначале, как найти ΔS тех или иных процессах.
1. Обратимый адиабатический процесс. Поскольку на всех стадиях такого процесса
а значит, в обратимых адиабатических процессах энтропия системы не меняется.
2. Нагревание системы.
а) Нагревание при постоянном объеме или давлении — термодинамически обратимый процесс. Поэтому в формуле (3.9) в качестве δQобр (точнее, теперь уже dQобр) следует взять теплоту реального процесса, которую, в свою очередь, можно найти через теплоемкость.
б) Действительно, исходя из формул (2.20–2.22), для произвольного количества вещества имеем:
Если полагать величину CP на рассматриваемом температурном интервале постоянной, то подстановка в (3.9) даёт:
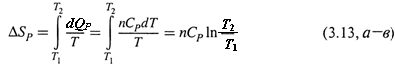 |
в) Аналогично, для нагревания воды при постоянном объёме
 |
г) Для жидких и твердых веществ CP = CV, поэтому для них
д) Из формул (3.13)—(3.14) видно, что
т. е. при нагревании степень энергетического беспорядка системы возрастает.
е) Пример. Теплоемкость воды —
Используя ее, по формуле (3.13, в) находим изменение энтропии 1 моля воды при его нагревании от 0°С до 100°С, т. е. от T1 = 273 К до Т 2 = 373 К:
3. Фазовые переходы.
а) Это тоже зачастую термодинамически обратимые процессы, которые при небольшом изменении условий могут сдвигаться в ту или иную сторону.
Для переходов в газ (ж↔г и тв↔г) это справедливо тогда, когда газ (пар) является насыщенным (что достигается обычно в закрытых сосудах). А для плавления (перехода тв↔ж) справедливо практически всегда.
б) Кроме того, в процессах типа ж↔г (при температуре кипения) и тв↔ж температура сохраняется постоянной; в процессах же типа тв↔г и ж↔г (при произвольной температуре) температуру поддерживают постоянной (для измерения теплоты перехода).
в) Поэтому в обоих случаях интегрирование не требуется.
Следовательно, ΔS опять-таки рассчитывается по теплоте процесса:
Пример 1. Теплота плавления льда такова:
Учитывая, что Тпл = 273 К, находим:

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 |
