


Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Там, где система координат соответствует порядку реакции, будет получаться
линейная зависимость.
б) Второй вариант — метод Вант–Гоффа.
I. По существу, необходимо определить показатель γ в уравнении
(Остальные реагенты, как было условлено, берутся в избытке, поэтому их концентрации почти не меняются и могут быть включены в константу скорости.)
 II. Прологарифмируем (17.47):
II. Прологарифмируем (17.47):
Получилась линейная зависимость lgυ от lgc. Причем, тангенс угла наклонаэтой зависимости равен искомому коэффициенту γ.
III. Следовательно, построив такой график (рис. 17.10) и определив угол его наклона, можно оценить порядок реакции по веществу.
3. Определение порядка по Т½.
а) Здесь проводится серия опытов: берут разные начальные концентрации исследуемого вещества и определяют время, за которое концентрация уменьшается вдвое. Критерий здесь — как Т½ зависит от начальной концентрации (c0).
б) Как мы знаем, для реакций разных порядков эта зависимость такова.
1-й порядок 2-й порядок 3-й порядок
Т½ ≠ f (c0) Т½ ~ 1/ c0 Т½ ~ 1/ (c0) 2
На этом завершим рассмотрение формальной кинетики простейших реакций.
В следующей главе мы обратимся ко второй проблеме кинетики – механизму химических реакций, но, конечно, – в самом общем смысле, поскольку конкретные механизмы реакций весьма многообразны.
Краткое содержание главы 17
В главе рассматривались КИНЕТИЧЕСКИЕ УРАВНЕНИЯ простейших реакций. Результаты можно свести в следующую таблицу.
 |
Глава 18. ПРИРОДА КОНСТАНТЫ СКОРОСТИ
В п. 16.4 мы говорили, что константа скорости зависит, во-первых, от специфики самой реакции (природы реагентов и конкретного механизма реакции), а во-вторых, от температуры. Влияние этих факторов на константу скорости и будет рассмотрено в данной главе.
18.1. Распределение частиц по скоростям и энергии
а) Первый фактор – специфика реакции – количественно отражается в энергии активации. В качестве последней используют две несколько отличающиеся величины:
- ΔGак – энергию Гиббса активации и
- Еак – энергию активации по Аррениусу, или просто энергию активации.
б) В трактовке обеих величин ключевую роль играет представление о том, что молекулы одного и того же вещества существенно различаются по энергии.
в) Поэтому рассмотрим вначале, как образуется такое распределение. Для простоты будем пока иметь в виду лишь кинетическую энергию молекул.
1. Распределение по проекциям скорости на направление. Мгновенную скорость (u) любой молекулы можно разложить на составляющие по направлениям – ux, uy, uz.
Так вот, прежде всего молекулы различаются по проекциям своей скорости на каждое направление; причём эти проекции постоянно меняются. Тем не менее общий характер распределения проекций остаётся постоянным и характеризуется двумя функциями.
а) F(ux) – интегральная функция распределения, т. е. вероятность того, что проекция скорости – не более значения ux. Очевидно, с ростом аргумента ux указанная вероятность возрастает от 0 до 1 (рис. 18.1,а).
 |
 б) Но обычно рассматривают не саму функцию F(ux), а её производную – плотность вероятности:
б) Но обычно рассматривают не саму функцию F(ux), а её производную – плотность вероятности:
Очевидно, ω(ux)∙dux – вероятность того, что проекция скорости про-извольной молекулы изуча-емой системы лежит в интервале dux, примыкаю-щем к точке ux.
I. Распределение проекций их является нормальным. Это значит, что оно
описывается следующей формулой:
II. Центр этого распределения — в нуле (рис. 18.1, б). Действительно, так как частицы с равной вероятностью могут двигаться в обоих направлениях оси х, то
средняя скорость по этой оси равна нулю. Величина
отклонение. Можно доказать, что
где М — молярная масса вещества.
2. Распределение частиц по абсолютной скорости (распределение Максвелла). А как найти распределение частиц по абсолютной скорости и?

а) Величина ω(ux,uy,uz) — это плотность вероятности того, что проекции скорости частицы равны ux,uy,uz (рис. 18.2). Она получается путем перемножения выражений вида (18.2):
б) Но некоторое значение скорости и может складываться из большого множества различных комбинаций значений ux,uy и uz. Величина этого множества определяется (в пространстве скоростей) площадью сферы радиуса и, равной 4πu2. Учитывая это, приходим к распределению частиц по абсолютной скорости:
 |
в) Подставим сюда формулы (18.2)—(18.4), учитывая 3 обстоятельства:
В последнем соотношении N — число частиц, а N0 — общее количество частиц. С помощью этого соотношения переходим от плотности вероятности к числу частиц, имеющих определенную скорость (точнее, к производной числа частиц по скорости).
г) В итоге получаем распределение Максвелла — распределение числа частиц по абсолютной скорости:

 |
В отличие от нормального распределения, здесь (рис.18.3) максимум распределения и средняя арифметическая скорость находятся не в нуле, а имеют положительные значения, которые, как можно доказать, таковы:
![]()
Ещё больше по величине значение среднеквадратичной скорости:
3. Распределение частиц по кинетической энергии.
а) Наконец, перейдём к распределению частиц по кинетической энергии. Для этого учтём следующее:

 б) Отсюда
б) Отсюда
в) Данное распределение похоже на предыдущее, но является более пологим (рис. 18.4), т. к. и степенная, и экспоненциальная зависимости здесь уже не такие сильные.
г) Исходя из формулы (18.10) или формулы (18.8,в), можно придти к известному выражению (1.2,а) для средней кинетической энергии идеального газа или идеального раствора (в расчёте на 1 моль вещества):
В частности, для 298 К получаем: Екср ≈ 3,7 кДж/моль.
18.2. Доля активных молекул и энергия активации
1. а) Итак, молекулы газообразного или растворённого вещества значительно различаются по энергии. Причём это касается не только кинетической энергии поступательного движения, но и энергии других видов движения – вращательного, колебательного, а также движения групп атомов в самих молекулах.
б) При этом молекулы постоянно сталкиваются друг с другом, с молекулами растворителя и прочих веществ. Поэтому в одних молекулах связи, которые должны разорваться в рассматриваемой реакции, оказываются более слабыми, в других – более сильными.
в) В итоге, можно говорить о неравномерном распределении не только механической, но и химической энергии.
2. а) Центральное представление химической кинетики состоит в том, что реакционноактивными являются не все молекулы (тогда бы реакция проходила мгновенно), а лишь те частицы, чья энергия не меньше (по модулю) некоего уровня, называемого энергетическим барьером, Еб.
Доля таких активных частиц соответствует на рис.18.4 площади заштрихованной области под кривой распределения.
б) Разница же между средней и барьерной энергией реагентов и есть энергия активации:
Заметим, что энергия активации – всегда положительная величина.
3. а) Таким образом, Еак показывает, сколько энергии надо сообщить 1 молю средних молекул, чтобы они все стали реакционноактивными.
б) Причём ясно: чем меньше Еак , тем больше доля активных молекул и, следовательно, выше скорость реакции.
в) То же самое можно сказать об энергии активации обратной реакции.
4. Какова же конкретная зависимость доли активных молекул (ζак) от энергии активации?
 |
а) Чтобы её определить, надо (как уже было сказано) найти площадь заштрихованной площади на рис. 18.4, т. е. рассчитать интеграл:
 |
б) Подстановка сюда (18.10) после ряда преобразований приводит к приближённому результату:
где L – некий множитель.
в) Таким образом, доля активных молекул с ростом энергии активации убывает практически по экспоненциальному закону. Это утверждение часто обозначается как закон Больцмана.
Заметим: на самом деле распределение Больцмана строго выводится для другой ситуации.
 5. а) Представление об энергии активации часто иллюстрируют также с помощью энергетической диаграммы (рис.18.5).
5. а) Представление об энергии активации часто иллюстрируют также с помощью энергетической диаграммы (рис.18.5).
Поскольку, как мы говорили, используют не только Еак , но и ΔGак, диаграмма дана для энергии Гиббса. В частности, энергия участников реакции (реагентов и продуктов) характеризуется через энергию Гиббса сгорания, ΔGсг (п.4.11). Но вида диаграммы это нисколько не меняет.
б) Итак, на рисунке ось абсцисс условно соответствует ходу реакции, а по оси ординат откладывается энергия. При этом показаны следующие величины.
I. ΔGср(реаг) и ΔGср(прод) – средние энергии (сгорания) реагентов и продуктов. Если в реакции – несколько реагентов, то ΔGср(реаг) – сумма их средних мольных энергий. То же – в отношении ΔGср(прод).
II. Разница между этими величинами – энергия реакции:
III. Энергетический барьер, ΔGб, является одинаковым для прямой и обратной реакций.
 |
IV. Энергии активации той и другой, соответственно, равны:
 |
V. Вычитая из первого уравнения второе, находим, что энергии активации прямой и обратной реакций тоже отличаются друг от друга на энергию реакции:
18.3. Теория активных столкновений
1. а) Мы выяснили, как зависит от энергии активации (при заданной температуре) доля активных молекул (18.14). Теперь надо вернуться к константе скорости и определить, как онá зависит от энергии активации, а также от температуры.
б) Есть два подхода к этим вопросам. Первый из них основан на теории активных столкновений.
2. Допустим, речь идёт о реакции вида:
а) С позиций названной теории считается: для того, чтобы субстраты прореагировали, необходимы три условия –
I. чтобы молекулы столкнулись,
II. чтобы они при этом имели правильную ориентацию друг относительно друга
III. и чтобы они обладали энергией выше барьерной.
 |
б) Этим условиям соответствуют три сомножителя в предлагаемой формуле скорости:
Здесь
I. v0 – скорость столкновений частиц (удары в секунду; деление на число Авогадро позволяет перейти к другим единицам – моль/с);
II. Р – т. н. стерический фактор (учитывает необходимость правильной ориентации);
III. ζак – уже известная нам доля активных молекул.
3. а) В свою очередь, очевидно, скорость столкновений частиц тем больше,
- чем больше их концентрация,
- чем выше скорость движения и
- чем больше размер молекул.
б) Расчёт, опускаемый нами, приводит к следующей формуле, учитывающей все эти влияния:
В ней nA и nB – концентрации молекул (количество частиц в единице объёма раствора), от которых легко перейти к молярным концентрациям cA и cB ;
uср – средняя скорость движения частиц, dАВ – их средний эффективный диаметр, A′ – константа.
 |
4. а) Подставим выражения (18.20,а-в) и (18.14) в формулу (18.19):
б) Всё, что стоит после концентраций, – это, очевидно, константа скорости. Поэтому получаем окончательное выражение:
в) Формула (18.22,а) – это очень известное уравнение Аррениуса, вначале предложенное Аррениусом на основании экспериментальных данных, т. е. эмпирически. А затем был осуществлён вывод этого уравнения, исходя из теории активных столкновений.
18.4. Анализ уравнения Аррениуса
1. а) Полученные выражения вскрывают те факторы, которые влияют на константу скорости.
Самый главный из них – энергия активации. С ростом энергии активации константа скорости убывает по экспоненте.
б) Кроме того, имеет значение и следующее: размер частиц (dАВ), их масса (от неё зависит скорость движения молекул, uср (18.8,б), стерический фактор Р, температура.
2. а) Какова же, согласно уравнению Аррениуса, зависимость k от температуры?
Поскольку в предэкпоненциальном множителе этого уравнения (18.22,б) температура не фигурирует, вся зависимость обусловлена температурой в показателе экспоненты.
 |
б) Можно убедиться, что с ростом Т константа скорости увеличивается. Действительно, производная k по Т – величина положительная:

в) Важная особенность: k не возрастает беспредельно, а стремится к максимальному уровню,
совпадающему с предэкспоненциальным множителем.
г) Отсюда — та S-образная зависимость k от Т, которая приведена на рис. 18.6.
 д) Однако чаще используют логарифмическую форму уравнения Аррениуса:
д) Однако чаще используют логарифмическую форму уравнения Аррениуса:
 |
Она удобна тем, что здесь имеется линейная зависимость между lnk и обратной температурой (1/Т).
3. С помощью уравнения Аррениуса по экспериментальным данным можно
найти энергию активации.
а) Первый способ — графический. Здесь определяют значения k при разных
температурах, строят график зависимости ln k от 1/Т и находят тангенс угла наклона этой линии (рис. 18.7):
б) Второй способ — аналитический. Здесь достаточно знать k всего при двух температурах. Подход — точно такой же, как при расчете стандартной энтальпии
реакции, когда известна константа равновесия при двух температурах (п. 5.9).
I. Действительно, составим систему уравнений:
 |
II. Вычитая из одного другое, приходим к следующему уравнению:
 |
(Ср. с (5.19).) Из него, зная k1, k2, T1 и Т2, нетрудно найти Еак.
 |
Кроме того, данное уравнение позволяет объяснить и правило Вант-Гоффа.
Суть этого правила в том, что обычно при повышении температуры на 10°
скорость реакции увеличивается в 2—4 раза:
б) Величина γ (показывающая, во сколько раз возрастает k при повышении
Т на 10°) называется температурным коэффициентом константы скорости.
Подставим в (18.27) Т2 = Т1+10. Тогда, с учетом сделанного определения, находим:
в) Пусть Т1 ≈ 300 К. В этом случае
- при подстановке γ = 2 получаем: Еак ≈ 50 кДж/моль,
- а при подстановке γ = 4 – Еак ≈ 100 кДж/моль.
Это означает, что правило Вант-Гоффа в области обычных температур справедливо для таких реакций, у которых энергия активации находится в пределах 50 ÷ 100 кДж/моль.
18.5. Теория активированного комплекса
Более общей, по сравнению с Еак, является ΔGак – энергия Гиббса активации. Эта величина вводится в рассмотрение во второй теории – теории активированного комплекса, разработанной Эйрингом.
1. Пусть опять речь идет о бисубстратной реакции. Согласно рассматриваемой теории, реакция проходит в две стадии:
 |
а) Первая стадия – обратимая; при этом те молекулы, которые обладают достаточной энергией, образуют активный комплекс.
Энергия данного превращения – это и есть энергия Гиббса активации.
Причём, как и для всякой другой обратимой реакции, стандартное значение этой энергии связано с константой равновесия формулой вида (4.35):
б) Вторая стадия процесса (18.30) – необратимый распад активированного комплекса на продукты; скоростью этого распада и определяется скорость реакции:
2. а) Обычно считается, что скорость второй стадии относительно невелика (по сравнению со скоростями прямой и обратной реакций первой стадии), так что на первой стадии практически успевает установиться равновесие.
б) Тогда можно выразить концентрацию активированного комплекса через концентрации исходных реагентов:
и подстановка в (18.32) даёт:
в) Сопоставление последней формулы с уравнением скорости исходной реакции
приводит к заключению:
Как видно, оно состоит в том, что константа скорости рассматриваемой реакции равна произведению соответствующих констант, характеризующих промежуточные стадии.
3. а) При этом ![]() — это, по сути дела, частота распада возбужденных комплексов с образованием продуктов — в расчете на 1 появляющийся комплекс (тогда это число комплексов, которые при такой частоте распались бы за 1 с), или в расчете на 1 моль комплексов (тогда это количество молей, распавшихся бы за 1 с).
— это, по сути дела, частота распада возбужденных комплексов с образованием продуктов — в расчете на 1 появляющийся комплекс (тогда это число комплексов, которые при такой частоте распались бы за 1 с), или в расчете на 1 моль комплексов (тогда это количество молей, распавшихся бы за 1 с).
б) Поскольку в комплексе соответствующая связь уже полностью готова к разрыву, она разорвется при первом же колебании, имеющем среднюю для данной температуры энергию. Следовательно, ![]() совпадает с частотой колебаний:
совпадает с частотой колебаний:
в) Данную частоту можно найти, если считать, что колебательная энергия комплекса равна его потенциальной энергии:
г) Тогда
Здесь фигурируют постоянные Больцмана (kБ) и Планка (h); вспомним их значения:
д) Подставляя их в (18.36, б), можно найти:
е) Отсюда следует, что образовавшийся комплекс перед превращением в продукт существует в среднем около
 |
4. а) Подставим выражения для констант
б) Она тоже (как и уравнение Аррениуса в предыдущей теории) вскрывает природу константы скорости и устанавливает ее зависимость от температуры.
в) Нетрудно убедиться: с точки зрения размерности эта формула верна лишь
для реакций первого порядка (где [k] = l/с). Для реакций же других порядков
в формулу Эйринга необходимо вводить дополнительный размерный множитель.
Например, в случае реакций второго порядка, где [k] = l/М·с (табл. 16.3), это
 |
множитель l/М:
18.6. Анализ уравнения Эйринга
1. а) Как и любая энергия Гиббса, энергия Гиббса активации выражается через энтальпию активации и энтропию активации:
б) Заметим: в результате образования комплекса упорядоченность расположения частиц повышается, так что энтропия снижается.
в) А теплота, в расчете на моль реагирующих частиц, поглощается (поскольку активный комплекс обладает повышенной внутренней энергией).
г) Следовательно, знаки величин таковы:
 |
2. Подстановка (18.42) в формулу Эйринга (18.40) дает:
а) Получается, что температура теперь входит не только в показатель экспоненты (как в уравнении Аррениуса), но и в предэкспоненциальный множитель.
б) И действительно, нагревание оказывает двоякое действие:
- во-первых, по принципу Ле-Шателье, она сдвигает равновесие первой (эндотермической) стадии процесса в сторону образования активированного комплекса,
- во-вторых, ускоряет распад этого комплекса.
 в) В итоге получается важное отличие от формулы Аррениуса: при высоких температурах — не стремление k к некоему пределу, а неограниченный рост (рис. 18.8):
в) В итоге получается важное отличие от формулы Аррениуса: при высоких температурах — не стремление k к некоему пределу, а неограниченный рост (рис. 18.8):
3. Какая же зависимость более правильная — по уравнению Аррениуса или по формуле Эйринга?
а) При относительно небольших температурах более точной, видимо, является формула Эйринга.
б) А при высоких температурах она перестает быть справедливой: рост константы скорости ограничивается тем или иным уровнем.
4. Наконец, установим связь величины ![]() (или ее компонентов) с энергией активации по Аррениусу (Eак).
(или ее компонентов) с энергией активации по Аррениусу (Eак).
 |
а) Для этого выразим константу скорости двумя способами — через уравнение Аррениуса и через формулу Эйринга:
б) Прологарифмируем оба выражения:
в) И, чтобы освободиться от разных констант (А и В), продифференцируем обе части равенства по T:
г) Отсюда и следует искомая связь:
д) Как видно, энергия активации по Аррениусу почти совпадает с энтальпией
(или теплотой) активации.
Таким образом, энергия Гиббса активации — более общее понятие: кроме энтальпии активации, она учитывает и изменение энтропии при активации.
е) Заметим: более точные расчеты показывают, что для бимолекулярной ре-
акции поправка в формуле (18.49) равна не RT, а 2RT.
Краткое содержание главы 18
В главе была рассмотрена ПРИРОДА КОНСТАНТЫ СКОРОСТИ.
1. ТЕОРИЯ АКТИВНЫХ СТОЛКНОВЕНИЙ исходит из следующего.
а) Молекулы вещества различаются по абсолютной скорости, образуя так называемое РАСПРЕДЕЛЕНИЕ МАКСВЕЛЛА, из которого вытекает распределение и по КИНЕТИЧЕСКОЙ ЭНЕРГИИ.
б) В реакцию же могут вступать лишь молекулы с энергией выше некоего барьерного уровня (Еб).
в) Разность между этим уровнем и средней энергией молекул вещества – ЭНЕРГИЯ АКТИВАЦИИ по Аррениусу: Еак = Еб – Еср .
г) Доля реакционноактивных молекул с ростом Еак убывает по экспоненте (ЗАКОН БОЛЬЦМАНА).
д) Кроме того, теория учитывает частоту столкновений молекул друг с другом, необходимость их правильной ориентации и ряд других факторов.
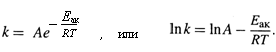 |
е) Это приводит к УРАВНЕНИЮ АРРЕНИУСА:
ж) Согласно ему, с ростом Т константа k растёт тоже, стремясь к предельному уровню А. Причём, если 50 < Eак < 100 кДж /моль, то при увеличении Т на 10 градусов k возрастает в 2–4 раза (ПРАВИЛО ВАНТ-ГОФФА).
з) Логарифмическая форма уравнения описывает линейную зависимость lnk от 1/T. Из неё следует: определив k при двух температурах, можно оценить и Еак, исходя из уравнения:
2. а) В ТЕОРИИ же ЭЙРИНГА считается, что молекулы, обладающие достаточной энергией, образуют вначале АКТИВНЫЙ КОМПЛЕКС.
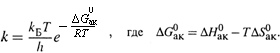 |
б) Расчет приводит к ФОРМУЛЕ ЭЙРИНГА:
в) Эта формула описывает БЕСПРЕДЕЛЬНЫЙ рост k при нагревании. Она предпочтительней при не очень больших температурах.
г) Энергии активации двух уравнений связаны соотношением:
![]()
Глава 19. КИНЕТИКА СЛОЖНЫХ ПРОЦЕССОВ
В данной главе будет рассмотрена формальная кинетика сложных процессов, под которыми понимают процессы, содержащие более одной стадии. Вообще говоря, один вид таких процессов мы уже обсуждали в п. 17.2, где речь шла об обратимых реакциях первого порядка
Их можно представить как двустадийные процессы:
Теперь рассмотрим еще несколько видов сложных (в основном, двустадийных) процессов.
19.1. Две параллельные реакции первого порядка

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 |
