


Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
б) Для смесей же с отрицательным отклонением диаграммы выглядят, как показано на рис. 9.7,а–б.
в) Еще раз заметим, что на диаграммах давления линия ж всегда идет выше, чем линия n, а на диаграммах кипения — ниже.
г) При наличии того или иного отклонения от идеальности диаграммы строят
только по экспериментальным данным. Теоретический расчет с использованием выкладок (9.7 и 9.12) затруднен из-за того, что аналитическая зависимость коэффициентов активности от концентраций обычно не известна.
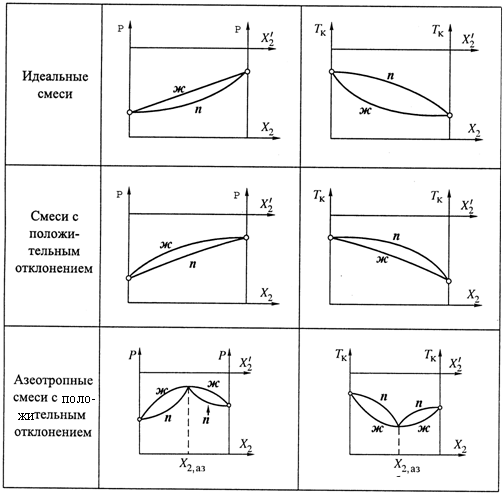
9.5. Азеотропные смеси
1. На рис. 9.2 мы приводили зависимость состава насыщенного пара (![]() ) от
) от
состава жидкой смеси (X2). Кривая этой зависимости идет с одной стороны от диагонали единичного квадрата, т. е. при любом составе смеси компонент 2 является более летучим, чем компонент 1. Это справедливо и для идеальных, и для многих реальных смесей.
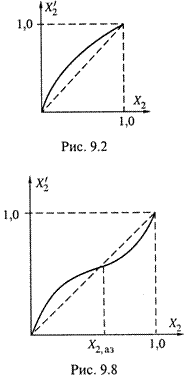 2. Но существуют так называемые азеотропные
2. Но существуют так называемые азеотропные
смеси, у которых график подобной зависимости в какой-то точке (X2,аз) пересекает диагональ квадрата (рис. 9.8). Это означает, что
- до указанной точки компонент 2 — более летучий (![]() ),
),
- после нее — менее летучий, а его содержание в паре становится меньше, чем в жидкости (![]() < X2).
< X2).
В самой же азеотропной точке летучесть обоих компонентов совершенно одинакова — состав пара совпадает с составом жидкой фазы.
3. Диаграммы давления и кипения для азеотропных смесей имеют характерный вид. Приведем их вначале для случая, показанного на рис. 9.8, т. е. для ситуации, когда при переходе через точку X2,аз вещество 2 из более летучего становится менее летучим.
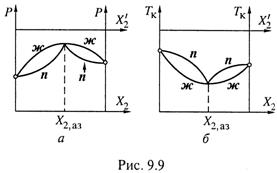 |
а) На диаграмме давления (рис. 9.9,а) для кривой ж мы получаем не просто
положительное отклонение от линейной зависимости (как на рис. 9.5), а и
наличие максимума в точке X2,аз.
I. Действительно, до этой точки компонент 2 — более летучий, отчего увеличение его содержания в жидкой смеси (![]() ) повышает общее давление пара над ней.
) повышает общее давление пара над ней.
II. После точки X2,аз данный компонент — уже менее летучий, и дальнейшее увеличение его относительного содержания в жидкой фазе теперь понижает общее давление пара.
III. А поскольку в точке X2,аз состав пара и жидкости совпадает, кривая п должна в этой точке сходиться с кривой ж. Отсюда на диаграмме — как бы две петли.
б) На диаграмме же температур кипения (рис. 9.9,6), соответственно, наблюдается минимум для кривой ж в азеотропной точке. Т. е. здесь — самая низкая температура кипения смеси. Линия п образует две дуги над линией ж.
4. Реже встречаются азеотропные смеси с отрицательным отклонением. Они характеризуются минимумом на диаграмме давления и максимумом на диаграмме кипения (рис. 9.10).
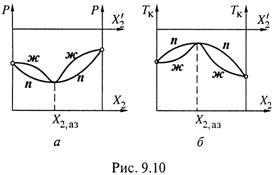 а) Заметим, что, подобно предыдущему случаю, линии ж на обеих диаграммах являются гладкими, т. е. не имеют в точке экстремума разрыва производной, как нередко их изображают.
а) Заметим, что, подобно предыдущему случаю, линии ж на обеих диаграммах являются гладкими, т. е. не имеют в точке экстремума разрыва производной, как нередко их изображают.
б) Линии же n находятся с выпуклой стороны линий ж, но при этом не могут заходить за экстремальные значения Р и Tk.
5. а) Однако каким бы ни был тип отклонения азеотропных смесей, им присуще уже известное нам свойство: в точках экстремума на диаграммах давления и кипения составы жидкой и газовой фаз смеси одинаковы.
Это утверждение обозначается как второй закон Коновалова. (Первый закон Коновалова был сформулирован в п.9.1.)
б) Классический пример азеотропной смеси с положительным отклонением — смесь H2O–спирт. Для данной системы азеотропная точка (массовая доля спирта) такова:
в) Примеры азеотропных смесей с отрицательным отклонением — водные растворы HCl и HNO3
9.6. Разделение жидкостей — перегонка и ректификация
а) И проведение научных исследований, и практика постоянно требуют разделения смеси из двух хорошо растворимых друг в друге жидкостей на отдельные компоненты. Используемый для этого метод основан на том, что, если жидкости различаются по летучести, при кипении пар обогащен более летучим компонентом.
б) Способность смеси к разделению характеризуется так называемым коэффициентом разделения α, показывающим, во сколько раз отношение компонентов в газовой фазе отличается от такового в жидкой фазе:
 |
Подставляя сюда формулы вида (9.4), нетрудно найти:
в) Значит, в идеальном случае коэффициент разделения не зависит от состава смеси и определяется только отношением давлений насыщенного пара над чистыми жидкостями. Чем сильней различаются эти давления, тем лучше разделяются жидкости. В случае неидеальных смесей коэффициент разделения зависит и от состава смеси, т. е. меняется по ходу изменения состава.
г) Известны три модификации данного метода — простая перегонка, фракционная перегонка и ректификация.
1-2. Простая и фракционная перегонка. Ее суть показана на рис. 9.11.
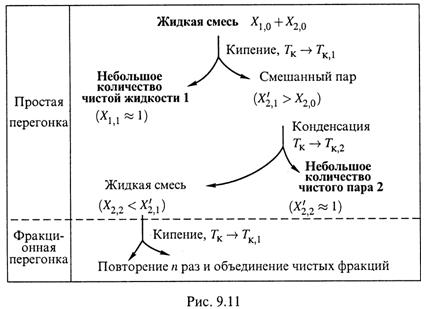 |
а) I. Вначале исходную смесь доводят до кипения и длительное время выпаривают, а образующийся пар отводят из системы. Остающаяся жидкая фаза обогащается менее летучим компонентом 1, отчего ее температура кипения постепенно повышается.
II. Если бы пар не отводился из системы, то, как уже отмечалось при обсуждении рис.9.4, после испарения всей жидкости состав газовой фазы достиг бы исходного состава жидкой фазы (Х2,0). Обогащения системы каким-либо компонентом в таком случае не происходило бы.
III. Благодаря же отведению пара общее содержание компонента 2 в остающейся системе не сохраняется постоянным, а понижается, и это позволяет достичь в жидкой фазе предельно низких значений Х2.
Следовательно, в результате данной процедуры получается небольшое количество почти чистого компонента 1 (в оставшейся жидкости).
б) I. Затем отводимый пар подвергают конденсации. В большей степени конденсируется опять-таки менее летучий компонент 1.
II. Если конденсат сразу отводить от пара, то в системе пар—образующаяся жидкость будет повышаться общее содержание компонента 2, что в итоге
позволит получить небольшое количество почти чистого пара этого компонента.
в) Таким образом, путем простой (однократной) перегонки и конденсации удается получить небольшое количество каждого компонента в практически
чистом виде.
 г) Но основная масса компонентов все же остается пока в жидкой смеси, образующейся при конденсации. Для увеличения выхода чистых веществ следует
г) Но основная масса компонентов все же остается пока в жидкой смеси, образующейся при конденсации. Для увеличения выхода чистых веществ следует
повторить указанную процедуру несколько раз и объединить чистые фракции. Такая более сложная модификация метода обозначается как фракционная перегонка.
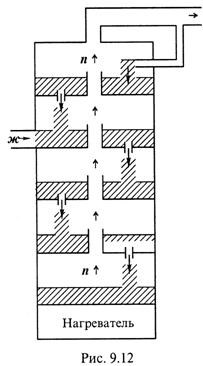
3. Ректификация. а) Третья разновидность метода – ректификация – представляет собой непрерывную фракционную перегонку, осуществляемую в специальных аппаратах – ректификационных колонках (рис.9.12). Колонка содержит систему «тарелок»; внизу находится нагреватель.
б) Поступающая в колонку жидкость попадает на одну из верхних «тарелок» и быстро доводится до кипения. Уже здесь часть её испаряется, а часть (обогащённая компонентом 1) из-за вытеснения новыми порциями жидкости перетекает на нижележащую «тарелку». Таким образом, жидкая фаза постепенно перемещается вниз, всё более обогащаясь компонентом 1.
в) Пар же, поднимаясь с каждой «тарелки», затем неоднократно конденсируется на вышележащих «тарелках», оставляя здесь менее летучий компонент 1.
г) В итоге, выходящий из колонки пар представлен почти только веществом 2, а вещество 1 остаётся в жидком состоянии.
4. Разделение азеотропных смесей. а) Если смесь — азеотропная, то методом перегонки можно получить в чистом виде только один компонент. Так, пусть речь идет об азеотропной смеси с положительным отклонением, т. е. на диаграмме кипения имеется локальный минимум (см. рис. 9.9,б).
б) На стадии испарения состав жидкой фазы меняется так, что изображающая точка на диаграмме движется от азеотропной точки. Это позволяет получить менее летучий компонент в чистом виде.
в) А при охлаждении пара изображающая точка уже движется по направлению к азеотропной точке. Например, пусть ![]() . При этом составе более летучим является компонент 2. Следовательно, при конденсации пара содержание вещества 2 в нем увеличивается, и пар по составу постепенно приближается к азеотропному.
. При этом составе более летучим является компонент 2. Следовательно, при конденсации пара содержание вещества 2 в нем увеличивается, и пар по составу постепенно приближается к азеотропному.
В этом же состоянии летучесть компонентов одинакова, что исключает
дальнейшее разделение.
г) По этой причине методом ректификации спирт очищают лишь до 96° (9.14, а). Одним из методов дальнейшей очистки азеотропной смеси является химическое связывание одного из компонентов. Например, воду связывают водоотнимающими веществами (СаСl2).
Краткое содержание главы 9
В главе рассмотрены СМЕСИ ДВУХ ЛЕТУЧИХ ЖИДКОСТЕЙ.
1. Общее давление пара над идеальной смесью линейно зависит от её состава:
Р0 = Р10 + Х2 (Р20 – Р10) ,
а по составу пар обогащён более летучим компонентом (ПЕРВЫЙ ЗАКОН КОНОВАЛОВА):
X2′ > X2 .
 |
2. а) Ключевые свойства смесей отражаются ДИАГРАММАМИ ДАВЛЕНИЯ И КИПЕНИЯ.
б) АЗЕОТРОПНЫЕ смеси имеют на диаграммах ЭКСТРЕМУМЫ, причём в этих точках составы жидкой и газовой фаз одинаковы (ВТОРОЙ ЗАКОН КОНОВАЛОВА).
3. Для разделения компонентов смеси используются 3 метода – простая перегонка, фракционная перегонка и ректификация. В случае азеотропных смесей эти методы позволяют получить в чистом виде лишь один компонент смеси.
Глава 10. СМЕСИ КОМПОНЕНТОВ, ОГРАНИЧЕННО
РАСТВОРИМЫХ ИЛИ НЕРАСТВОРИМЫХ ДРУГ
В ДРУГЕ В ЖИДКОМ ИЛИ В ТВЕРДОМ СОСТОЯНИИ
10.1. Ограниченно растворимые жидкости
1. До сих пор речь шла о смесях таких жидкостей, которые неограниченно
растворимы друг в друге. Но нередко растворимость ограничивается каким-то верхним пределом. Тогда, в зависимости от соотношения компонентов, система может находиться в виде не только одной, но и двух жидких фаз.
2. Это иллюстрируется схемой (рис. 10.1) для смеси H2O–анилин, где использованы обозначения: 1 — H2O, 2 — анилин, W2 — массовая доля анили-
на в смеси.
а) При небольшом содержании анилина (первый сосуд) получается
однофазный раствор анилина в воде (фаза I).
б) Если же содержание анилина превышает уровень растворимости (W2,I) (второй сосуд), то образуются 2 фазы:
I. насыщенный раствор анилина в воде с концентрацией W2,I,
II. насыщенный раствор воды в анилине с массовой долей анилина W2,II.
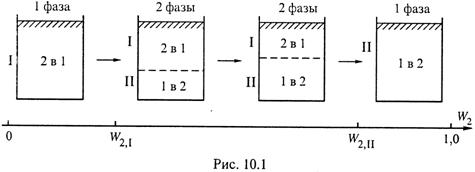 |
в) По мере увеличения содержания анилина в системе возрастает и доля фазы II (третий сосуд).
 г) Наконец, когда обшее содержание анилина становится выше W2,II, тогда фаза I перестает существовать, так как вся вода теперь может раствориться в анилине, т. е. остается только фаза II (четвертый сосуд).
г) Наконец, когда обшее содержание анилина становится выше W2,II, тогда фаза I перестает существовать, так как вся вода теперь может раствориться в анилине, т. е. остается только фаза II (четвертый сосуд).
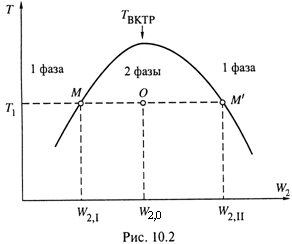
3. Но указанные значения растворимости — W2,I и W2,II — зависят от температуры. В данном случае эта зависимость выглядит так, как показано на рис. 10.2.
а) Пусть исходная температура — T1. Ей соответствуют значения растворимости W2,I и W2,II, так что в интервале W2,I < W2 < W2,II жидкость находится в двухфазном состоянии, а за пределами этого интервала — в однофазном.
б) При нагревании взаимная растворимость данных веществ повышается, отчего интервал двухфазности суживается.
в) И, наконец, при некоторой температуре TВКТР, называемой верхней критической точкой растворимости (отсюда – аббревиатура ВКТР), этот интервал обращается в ноль, т. е. при TВКТР смесь находится в однофазном состоянии при любом составе.
4. Рассмотрим распределение общей массы системы по фазам при температуре T1 с произвольным составом смеси W2,0.
а) Исходим из соотношения для массовой доли анилина в системе:
б) После элементарных преобразований находим массовую долю фазы I:
Таким образом, масса фазы I определяется на рис. 10.2 отрезком OM΄ (ограниченным значением W2,II), а масса фазы II, очевидно, отрезком ОМ (который
ограничен значением W2,I).
в) Это — очередное перекрёстное правило рычага (как и правило, сформулированное в п. 9.2 для диаграммы кипения идеальной смеси). Заметим, что существуют смеси и с другой зависимостью растворимости от температуры.
10.2. Взаимно нерастворимые жидкости
Теперь обратимся к смесям таких веществ, которые практически нераство-
римы друг в друге. Примеры — вода—масло, вода—бензол.
1. Подобные смеси имеют две принципиальные особенности.
а) Первая: так как и в смеси жидкости находятся в виде однородных мультимолекулярных структур (например, в виде капель из молекул одного типа), то присутствие одной жидкости не влияет на давление пара другой (в отличие от взаимно растворимых жидкостей). Т. е., независимо от состава смеси давление пара каждого компонента — то же, что и для чистого компонента, — ![]() и
и ![]() .
.
б) Но пузырьки пара, образующиеся при закипании смеси, содержат пары обоих веществ. Поэтому закипание наступает тогда, когда с внешним давлением
сравняется общее давление обоих паров: 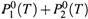 .
.
Отсюда вытекает вторая особенность: температура кипения смеси ниже
температуры кипения любого компонента
Для взаимно же растворимых жидкостей (исключая азеотропные смеси) температура кипения смеси, как видно из диаграммы рис. 9.4, находится в интервале
между температурами кипения отдельных компонентов:
2. Приведем конкретный пример. У воды и у скипидара давление паров
достигает атмосферного, соответственно, при следующих температурах:
а совокупное давление паров этих веществ сравнивается с атмосферным при
(в широком диапазоне состава смеси); т. е., действительно, смесь закипает при более низкой температуре, чем любой чистый компонент.
3. а) На указанной особенности основан метод очистки органических жидкостей от примесей — перегонка с водяным паром. Дело в том, что многие органические вещества при своей температуре кипения разлагаются. Поэтому их смешивают с водой. Температура кипения становится ниже, чем даже у воды, и органическое вещество уже выдерживает кипячение.
б) Последовательность операций такова. Вначале смесь испаряют (при этом
загрязняющие вещества в пар не переходят). Затем пар конденсируют; образующаяся жидкая фаза расслаивается на две фазы — воду и органическую
жидкость. Вторую фазу осторожно отделяют.
4. Каково соотношение компонентов смеси в паре?
а) Учтем, что испарение производят в некотором фиксированном (постоянном) объеме V. И запишем уравнение Клайперона—Менделеева для компонентов в газообразном состоянии:
Отсюда видно, что в образующемся паре количество компонента пропорционально давлению его насыщенного пара при данной температуре.
б) Кроме того, нетрудно найти массовую долю компонента в паре:
в) После подстановки ![]() и
и ![]() получаем:
получаем:
 |
Если 1 — H2O, а 2 — очищаемая жидкость, то видно: массовая доля этой жидкости в паре тем больше, чем больше отношение 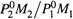 ..
..
10.3. Экстракция
1. Система взаимно нерастворимых жидкостей позволяет осуществить еще одну важную процедуру — экстракцию. Речь идет уже о трехкомпонентной системе: она включает две несмешивающиеся жидкости и распределенный между ними третий компонент (рис. 10.3).
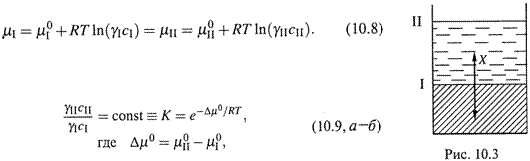 2. а) При равновесии химические потенциалы этого компонента в двух фазах одинаковы:
2. а) При равновесии химические потенциалы этого компонента в двух фазах одинаковы:
б) Отсюда
т. е. отношение активностей третьего компонента в фазах не зависит от общего количества этого компонента в фазах.
Здесь γi – коэффициенты активности, а через К обозначается термодинамическая константа распределения. Она, как любая константа равновесия, зависит от температуры и давления.
в) Отношение же концентраций называется коэффициентом распределения:
Коэффициент распределения зависит от концентраций, так как при их изменении могут меняться коэффициенты активности. В первую очередь это относится к электролитам.
3. Так вот, часто вещества распределяются между двумя несмешивающимися жидкостями очень неравномерно.
Это позволяет с помощью одной такой жидкости извлекать вещества из другой жидкости. Данная процедура и называется экстракцией; весьма широко она применяется в фармации. Например,
- с помощью водных сред из разных объектов извлекают водорастворимые компоненты,
- а с помощью неполярных жидкостей (например, масел, бензола, ацетона и т. д.) — соединения углеводородной природы.
4. Рассмотрим, от чего зависит эффективность экстракции.
а) Обозначим: VI — объем первой жидкости (где изначально находилось экстрагируемое вещество), VII — объем второй жидкости (с помощью которой осуществляется экстракция). И будем считать, что в обеих жидкостях коэффициенты активности вещества близки к единице: γI ≈ γII ≈ 1,0.
б) Тогда при равновесии
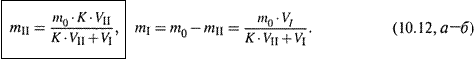 |
в) Отсюда находим массу вещества в каждой из фаз:
Видно: чем больше К и VII, тем больше масса экстрагированного вещества (mII).
5. а) Можно осуществить и так называемую дробную экстракцию — разделить объем VII на п равных частей: экстрагировать сначала одной частью и удалить фазу II, добавить очередную порцию экстрагента, вновь ее удалить — и т. д.
б) Тогда, например, в формулах второй экстракции вместо т0 следует ставить то количество вещества, которое осталось в фазе I после первой экстракции:
в) Обобщая последнюю формулу, получаем выражения для массы вещества в каждой из фаз после п экстракций:
 |
г) Если продифференцировать формулу для mI,n по п, можно убедиться:
Это значит, что при росте числа экстракций (при том же общем объеме экстрагента) количество вещества, остающегося в первой жидкости, становится всё меньше, т. е. дробная экстракция эффективней, чем простая.
10.4. Затвердевание бинарных жидких смесей:
простейший случай
Теперь обратимся к затвердеванию жидких смесей.
а) Для полноты картины вначале коротко остановимся на ситуации, ког-
да компоненты неограниченно взаимно растворимы и в жидком, и в твердом
состояниях. Обычно это вещества, близкие по своей природе, — например,
Ag–Au, NaCl–NaBr и т. д. Но в каждой паре одно из веществ (1) плавится (и затвердевает) при более высокой температуре, а другое (2) — при более
низкой.
 б) В идеальном случае диаграмма плавления (рис. 10.4) имеет точно такой же вид, как диаграмма кипения идеальной смеси (см. рис. 9.4), т. е. для смеси Tпл будет зависеть от состава смеси — уменьшаться с увеличением доли в жидкой фазе
б) В идеальном случае диаграмма плавления (рис. 10.4) имеет точно такой же вид, как диаграмма кипения идеальной смеси (см. рис. 9.4), т. е. для смеси Tпл будет зависеть от состава смеси — уменьшаться с увеличением доли в жидкой фазе ![]() легкоплавкого компонента. Это изображено верхней кривой диаграммы — линией ликвидуса.
легкоплавкого компонента. Это изображено верхней кривой диаграммы — линией ликвидуса.
в) В кристаллах, образующихся при охлаждении смеси до Tпл , тоже содержатся оба компонента (так как они взаиморастворимы и в твёрдом состоянии), но второго (более легкоплавкого) компонента меньше (X2 < X2′). Соответственно, можно построить и линию солидуса (нижняя линия на диаграмме): она связывает состав твёрдой фазы с Tпл.
г) Как и для перехода ![]() , здесь тоже возможны другие варианты диаграммы плавления, в т. ч. с экстремумами на кривых ликвидуса и солидуса. Например, для смеси Cu-Au наблюдается минимум на этих линиях.
, здесь тоже возможны другие варианты диаграммы плавления, в т. ч. с экстремумами на кривых ликвидуса и солидуса. Например, для смеси Cu-Au наблюдается минимум на этих линиях.
Следовательно, справедливо то, что говорилось об азеотропной смеси: в точке экстремума составы жидкой и твердой фаз одинаковы.
10.5. Затвердевание смесей, в которых компоненты взаимно нерастворимы в твердом состоянии
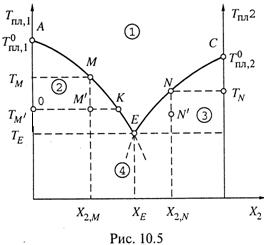 1. а) Бывают также смеси, где взаимная растворимость компонентов сохраняется только в жидком состоянии. При затвердевании таких смесей образуются не смешанные, а индивидуальные кристаллы. Примеры подобных смесей: Sb–Pb, H2O–соль (NaCl, CaCl2 и пр.), аспирин–амидопирин и пр.
1. а) Бывают также смеси, где взаимная растворимость компонентов сохраняется только в жидком состоянии. При затвердевании таких смесей образуются не смешанные, а индивидуальные кристаллы. Примеры подобных смесей: Sb–Pb, H2O–соль (NaCl, CaCl2 и пр.), аспирин–амидопирин и пр.
б) Диаграмма плавления (или затвердевания) (рис. 10.5) при этом имеет совсем иной вид, чем в предыдущем случае.
в) Чтобы её понять, вспомним одно из коллигативных свойств растворов — понижение температуры замерзания растворителя в присутствии растворенного вещества (п.8.6). Только теперь будут использованы термины «температура кристаллизации» или «температура плавления», что, по существу, одно и то же.
2. а) Так, пусть вначале в системе преобладает компонент 1, выступающий в качестве растворителя. Тогда при увеличении X2 температура кристаллизации вещества 1 понижается. Линия АЕ — это зависимость Tпл,1 от состава смеси.
б) Когда, например, при составе смеси X2,M система охлаждается до температуры TM, начинается кристаллизация вещества 1, а вещество 2 остается в жидкой фазе.
в) Доля вещества 2 в жидкой фазе постепенно возрастает, отчего Tпл,1
снижается еще больше — и идет движение в направлении точки Е.
3. а) Аналогично, при высоком содержании компонента 2 он рассматривается
уже как растворитель, и для него справедливо то же самое: при увеличении доли вещества 1 (т. е. при снижении X2) Tпл,2 снижается, чему и соответствует кривая СЕ.
б) При охлаждении системы до TN при составе X2,N начинается кристаллизация вещества 2; вещество же 1 остается в жидкой фазе, где его содержание постепенно возрастает.
в) И мы вновь двигаемся к точке Е, но уже с другой стороны, чем в предыдущем случае.
4. Естественно, что две указанные зависимости где-то должны пересекаться. Очевидно, в точке пересечения (Е),
- во-первых, температура плавления смеси является наименьшей (TE),
- а во-вторых, в кристаллическое состояние переходят сразу оба компонента, причем в той же пропорции, в какой они находятся в жидкой фазе.
Таким образом, в точке Е в равновесии находятся сразу три фазы: одна жидкая и две твердые — кристаллы одного и кристаллы другого компонента.
5. а) Линия АЕС вновь называется линией ликвидуса, так как в точках выше её
(область 1) система имеет только одну жидкую фазу.
б) Изотерма же TE — линия солидуса: в точках ниже ее (область 4) система имеет только твердые фазы.
в) Точка Е называется точкой эвтектики, а соответствующие ей состав смеси (XE) и температура (TE) — эвтектическими. Всего диаграмма имеет четыре области.
6. Применим к ним, а также к точкам, лежащим на ординатных осях (которые соответствуют условиям X2 = 0 или X1 = 0), правило фаз Гиббса:
а) Везде следует положить п = 1, так как из внешних факторов имеет значение только температура.
б) Кроме того, на ординатных осях К = 1 (в системе — только один компонент), а в остальных точках диаграммы К = 2.
в) Поэтому для точек на осях и для остальных точек выражение (6.18,б) соответственно преобразуется к виду:
7. Результаты применения этих выражений суммированы в табл. 10.1.
а) Например, для чистых компонентов (на ординатных осях, кроме точек А и С) С = 1, т. е. в некоторых пределах можно менять 1 параметр (температуру) без изменения фазового состояния системы — жидкого (выше точек А и С) или твердого.
б) В области 1 С=2, т. е. можно менять 2 параметра (и Т, и состав), сохраняя систему в жидком состоянии.
Т а б л и ц а 10.1
Ординантные оси (кроме точек А и С ) | Ф = 1 (ж или тв1) | С = 2 – 1 = 1 |
Точки А и С | Ф =2 (ж + тв1) | С = 2 – 2 = 0 |
Область 1 | Ф = 1 (ж) | С = 3 – 1 =2 |
Линия АЕ и область 2 | Ф = 2 (ж + тв1) | С = 3 – 2 = 1 |
Линия СЕ и область 3 | Ф = 2 (ж + тв2) | |
Точка Е | Ф = 3 (ж + тв1 + тв2) | С = 3 – 3 = 0 |
Область 4 | Ф = 2 (тв1 + тв2) | С = 3 – 2 = 1 |
в) Область 2. Пусть это точка М' на рис. 10.5; т. е. смесь состава X2,M находится при температуре TM' .

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 |
