


Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
б) Более конкретно это выглядит так:
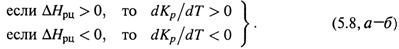
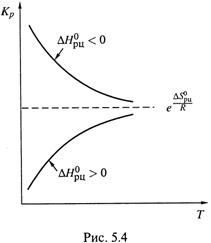
В самом деле, если прямое направление реакции – эндотермическое (ΔНрц > 0), то из-за сдвига реакции в этом направлении (под действием нагревания) в системе увеличивается доля продуктов – константа равновесия возрастает (dKр/dT > 0). Аналогично влияет на равновесие введение в систему иной энергии – не тепловой, а, например, световой, если система способна поглощать эту энергию.
Обсудим также количественную сторону влияния температуры на термодинамические параметры реакции – ΔSрц, ΔGрц и Кр .
5.8. Влияние температуры на ΔSрц и ΔGрц
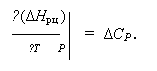 |
1. а) В п. 2.5 было показано, что зависимость энтальпии реакции от температуры в изобарных условиях определяется величиной ΔСР – разностью молярных теплоёмкостей участников:
б) Так, если теплоёмкость продуктов – выше, чем реагентов (ΔСР > 0), то при нагревании системы всё больше теплоты остаётся в продуктах, и реакция (если она эндотермическая) требует больше теплоты – ΔHрц возрастает.
в) Был рассмотрен также пример, который показывал, что фактическая зависимость ΔHрц от температуры – довольно слабая, отчего ею обычно пренебрегают.
2. Такие же свойства присущи и энтропии реакции, ΔSрц.
а) Действительно, для нагревания 1 моля произвольного участника реакции
можно записать:
![]()
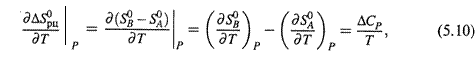 |
где S0 — абсолютная энтропия вещества, а CP — его изобарная теплоемкость.
б) Тогда для энтропии реакции (на примере простейшей реакции A → B) получаем:
 |
что дает уравнение
сходное с (2.29).
в) Таким образом, и для энтропии реакции температурная зависимость опре-
деляется разностью теплоемкостей продуктов и реагентов. Из-за малости
данной разности зависимостью ![]() от температуры обычно тоже можно пренебречь.
от температуры обычно тоже можно пренебречь.
3. а) Обратимся к стандартной энергии Гиббса реакции:
![]()
(Заметим: в данном случае температура считается не стандартной — 298 К, — а произвольной.) Полагая, что ![]() и
и ![]() не зависят от температуры, получаем:
не зависят от температуры, получаем:
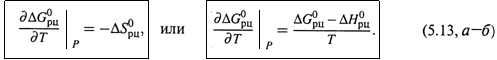 |
б) Второе из этих выражений называется уравнением Гиббса-Гельмголъца (это дифференциальное уравнение относительно ![]() ).
).
в) Однако в данном случае более наглядно первое уравнение. Из него видно, что производная ![]() по температуре равна энтропии реакции, взятой с обратным знаком.
по температуре равна энтропии реакции, взятой с обратным знаком.
г) Это выражение весьма близко к формуле (4.20,а):
устанавливающей, что температурная зависимость энергии Гиббса процесса
определяется энтропией системы. Но данная формула была выведена лишь для обратимых процессов.
 д) В отличие от этого, соотношение
д) В отличие от этого, соотношение
(5.13,а) справедливо для произвольных, в т. ч. и необратимых реакций. Правда, оно исходит из других упрощающих допущений —
независимости ![]() и
и ![]() от температуры.
от температуры.
е) Итак, согласно (5.13,а), характер из-
менения ![]() с ростом температуры
с ростом температуры
определяется знаком ![]() .
.
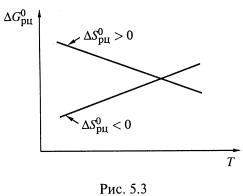
Если ![]() , то
, то ![]() снижается (или становится более отрицательным), т. е. способность реакции к самопроизвольному протеканию увеличивается.
снижается (или становится более отрицательным), т. е. способность реакции к самопроизвольному протеканию увеличивается.
А если ![]() , то зависимость
, то зависимость ![]() от температуры обратная (рис. 5.3).
от температуры обратная (рис. 5.3).
5.9. Зависимость Kp от температуры
1. Общий характер этой зависимости нам уже известен (5.8,а-б). Теперь
установим ее конкретный вид.
а) Выразим Kp из соотношения (4.35) и используем (5.12):
![]()
Видно, что, если ![]() и
и ![]() от температуры не зависят, то при росте температуры Kp стремится к некоторому предельному уровню (5.14,6), который при
от температуры не зависят, то при росте температуры Kp стремится к некоторому предельному уровню (5.14,6), который при 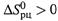 больше 1, а при
больше 1, а при ![]() — меньше 1.
— меньше 1.
б) Для определения характера зависимости Kp от Т (убывание или возраста-
ние) продифференцируем формулу (5.14, а):
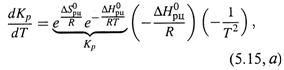
 |
откуда
Таким образом, знак производной ![]()
совпадает со знаком ![]() , что соответствует неравенствам (5.8,а-б) и показано на рис. 5.4. (Сравним: знак производной
, что соответствует неравенствам (5.8,а-б) и показано на рис. 5.4. (Сравним: знак производной ![]() определяется знаком
определяется знаком ![]() , но противоположен ему (5.13,а).)
, но противоположен ему (5.13,а).)
2. Уравнение, связывающее Kp с температурой, называют уравнением изобары химической реакции (или, наряду с уравнением изотермы химической реакции (4.34), — уравнением Вант-Гоффа). Его можно записать в разных формах.
 |
а) По сути дела, уже формула (5.14,а) является уравнением изобары. Но
обычно ее представляют в логарифмическом виде:
Чем полезно это выражение? Из него следует, что если
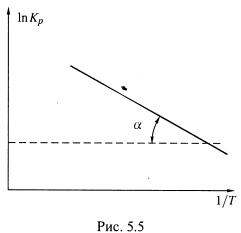 б) Следовательно, если экспериментально найти константу равновесия реакции
б) Следовательно, если экспериментально найти константу равновесия реакции
при нескольких температурах и построить график в указанных координатах, то по тангенсу угла наклона прямой можно найти энтальпию реакции.
Вместе с тем можно найти также стандартную энергию Гиббса (при какой-то температуре ![]() ) и энтропию реакции:
) и энтропию реакции:
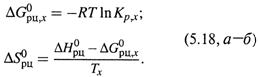
в) Причем, достаточно ограничиться всего двумя точками, т. е. измерить Kp при двух температурах — T1 и T2.
 |
Действительно, запишем уравнение (5.16) для температуры T1, затем — для
температуры T2 и вычтем одно уравнение из другого. Тогда член
Это еще одна форма уравнения изобары химической реакции. С ее помощью,
зная Kp1 и Kp2 (при температурах T1 и T2), нетрудно рассчитать ![]() .…
.…
 |
3. Наиболее общей формой уравнения изобары считается уравнение (5.15,6), если в нем перенести Kp из правой части в левую и взять под знак дифференциала (то же получается при дифференцировании формулы (5.16)):
Здесь в самом сжатом виде определена температурная зависимость Kp.
Краткое содержание главы 5
Глава посвящена ХИМИЧЕСКОМУ РАВНОВЕСИЮ.
1. Так, в ней сформулированы законы, касающиеся химического равновесия.
а) ЗАКОН ДЕЙСТВУЮЩИХ МАСС – отношение концентраций при равновесии не зависит от их исходных значений:
б) ЗАКОН ГЕССА ДЛЯ КОНСТАНТ РАВНОВЕСИЯ – для альтернативных цепей реакций равны произведения констант равновесия:
в) ПРИНЦИП ЛЕ ШАТЕЛЬЕ — в ответ на некоторое возмущение (изменение концентраций, общего давления или температуры) в обратимой реакции усиливается одно из двух направлений — и результат внешнего воздействия частично ослабевает.
2. Кроме того, мы установили количественную ЗАВИСИМОСТЬ ОТ ТЕМПЕРАТУРЫ термодинамических параметров реакции.
а) В случае ЭНТРОПИИ зависимость является слабой (из-за малости ![]() ), а в случае ЭНЕРГИИ ГИББСА — почти линейной:
), а в случае ЭНЕРГИИ ГИББСА — почти линейной:
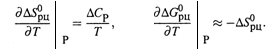 |
б) Для КОНСТАНТЫ же РАВНОВЕСИЯ получена серия соотношений, из которых наиболее важны следующие:
 Таким образом, зная Кр при двух температурах, можно найти ΔНорц.
Таким образом, зная Кр при двух температурах, можно найти ΔНорц.
ЗАДАЧИ К РАЗДЕЛУ 1
1. При t = 180° С 4 г водорода занимают объём V1 = 3л. Определить работу при изотермическом расширении этого количества водорода до объёма V2 = 4,8 л.
Решение
Имеется n = m/M(H2) = 2 моля водорода. Искомая работа, согласно (1.6) и с учётом того, что R ≈ 8,31 Дж/(моль∙К), а Т = 453 К, такова:
w = – nRT ln(V2 /V1) = 3,53 кДж. (I.1)
2. Рассчитать разность теплот QP – QV (на 1 моль ацетальдегида или на 1 моль этанола) при Т1 = 298 К и (отдельно) при Т2 = 400 К для реакции
Ацетальдегид (г) + H2 (г) = Этанол (при Т1 – ж; при Т2 – г) .
Решение
а) Исходя из (1.10), (1.11) и (2.5), разность теплот изобарного (QP) и изохорного (QV) проведения реакции равна работе расширения (или сжатия) газа:
QP – QV = –wP = PΔV = ΔnгазRT , (I.2)
где Δnгаз – изменение количества газообразных веществ в системе.
б) В расчёте на 1 моль реагирующего ацетальдегида или на 1 моль образующегося этанола, количество газообразных веществ снижается при Т1 на 2 моля (Δnгаз = –2), а при Т2 – на 1 моль (Δnгаз = –1).
в) В итоге, подстановка величин в (I.2) даёт: искомая разность теплот при Т1 равна –4,95 кДж/моль, а при Т2 – –3,32 кДж/моль.
3. Для метана ΔНообр = –74,85 кДж/моль. Определить ΔЕообр метана.
Решение
а) Реакция образования метана: 2Н2 + С = СН4 .
В расчёте на 1 моль образующегося метана, Δnгаз = –1.
б) Исходя из (2.8) и учитывая, что Т =298 К, получаем:
ΔEообр = ΔНообр – PΔV = ΔНообр – ΔnгазRT = –72,37 кДж/моль . (I.3)
4. Найти ΔE при испарении m = 0,2 кг этанола при температуре кипения (tк = 78,3°С). Теплота парообразования при той же температуре – Q1 кг = 856,9 кДж/кг. Изменением объёма жидкой фазы спирта пренебречь.
Решение
а) Согласно (1.7),
ΔE = m(Q1 кг + w) = m(Q1 – PΔV1 кг), (I.4)
где ΔV1 кг – увеличение парциального объёма, занимаемого испаряющимися молекулами (в расчёте на 1 кг вещества).
б) Перейдём к ΔV1 моль (изменению объёма в расчёте на 1 моль вещества):
ΔV1 кг = ΔV1 моль / M ; (I.5)
здесь М – молярная масса этанола.
в) Тогда, в соответствии с (2.5),
PΔV1 моль = ΔnгазRT, где Δnгаз =1,
откуда
ΔE = m(Q1 – RT / M) ≈ 159 кДж. (I.6)
Здесь учтено, что Тк = 351,3 К и М = 46 г/моль = 0,046 кг/моль.
5. Расcчитать ΔН нижеследующей реакции при Т = 500 К, используя значения ΔНообр и СР её реагентов и продуктов:
2NaBr (кр) + Сl2 (г) = 2NaCl (кр) + Br2 (ж)
ΔНообр, кДж/моль -
СР, Дж/(моль∙К) 88,3 33,9 50,9 75,7
Решение
а) В соответствии с формулой (2.18,а), при стандартной температуре (25оС, или 298 К) энтальпия реакции равна
ΔНорц = 2 ΔНообр (NaCl) – 2 ΔНообр (NaBr) = – 92 кДж/моль. (I.7)
б) Находим также разность теплоёмкостей продуктов и реагентов (2.27):
ΔСР = 2 СР (NaCl) + СР (Br2) – 2 СР (NaBr) – СР (Cl2) = –33 Дж/(моль∙К) . (I.8)
в) Энтальпию реакции при указанной в задаче температуре (для которой ΔТ = 202 К) оцениваем по формуле Кирхгофа (2.27):
ΔНрц (500 K) ≈ ΔНорц + ΔСР∙ΔТ ≈ – 99 кДж/моль (I.9)
6. Найти ΔS для превращения m = 1 г жидкой воды при Т1 = 273 К в пар при Т2 = 423 К. Давление считать нормальным: Р = 1 атм = 101,3 кПа (1.3). Теплоёмкость жидкой воды – СР(ж) ≈ 4,18 Дж/(г∙К) (2.26), а водяного пара – СР(г) ≈ 1,99 Дж/(г∙К). Теплота испарения при температуре кипения – ΔНисп = 40 кДж/ моль (3.18).
Решение
а) Последнюю величину отнесём к 1г воды:
ΔНисп = 40 кДж/моль : 18 г/моль = 2,25 кДж/г (I.10)
б) По аналогии с формулой (3.19), результирующее изменение энтропии рассчитывается как сумма ΔSi трёх процессов:
- нагревания жидкой воды от 0 до 100оС (т. е. от 273 К до 373 К),
- испарения воды при 100оС (373 К)
- и нагревания водяного пара от 100оС до 150оС (от 373 К до 423 К):
Ткип Т2
ΔS = СР(ж) ∫ dT / T + ΔНисп /Tкип + СР(г) ∫ dT / T = (I.11,a)
Т1 Ткип
= СР(ж) ln(Tкип /T1 ) + ΔНисп /Tкип + СР(г) ln(T2 /Tкип ) ≈ 7,59 Дж/(г∙К) . (I.11,б)
7. Рассчитать ΔS после смешивания двух порций спирта (m1 = 2,3 г, t1 = 70оC; m2 = 5,8 г, t2 = 10оC) и выравнивания их температур (до tср). Молярная теплоёмкость спирта – С = 111,4 Дж/(моль∙К). Изменением объёма системы пренебречь.
Решение
а) Количество теплоты, отданной первой (более горячей) порцией, равно по абсолютной величине количеству теплоты, полученной второй порцией спирта:
–Q1 = Q2 , или, согласно (2.20,а), m1С (t1 – tср) = m2С (tср – t2) . (I.12)
Отсюда
tср = (m1 t1 + m2 t2 ) /(m1 + m2) = 27oC; Тср = 300 K. (I.13)
б) Изменение энтропии каждой порции, согласно (3.14), таково:
ΔS1 = n1 ∙С∙ ln(Tср /T1) = (m1/M) ∙C∙ln(Tср /T1) = –0,73 Дж/К, (I.14,a)
ΔS2 = n2 ∙С∙ ln(Tср /T2) = (m2/M) ∙C∙ ln(Tср /T2) = +0,97 Дж/К, (I.14,б)
где М = 46 г/моль – молярная масса этанола, Т1 = 343 К, Т2 = 283 К. Энтропия остывающей порции уменьшается, а нагревающейся порции – возрастает.
в) Результирующее изменение энтропии системы – положительная величина:
ΔS = ΔS1 + ΔS2 = +0,24 Дж/K . (I.15)
Это иллюстрирует второе начало термодинамики в отношении процессов теплопередачи. – Итогом таких процессов является не только переход теплоты от более тёплого объекта к более холодному (что выравнивает температуры), но и возрастание энтропии системы в целом.
8. Вычислить ΔGо для следующей реакции (при 25оС), используя нижеприведённые термодинамические данные:
СО2 (г) + 4 Н2 (г) = СН4 (г) + 2 Н2О (ж)
ΔНообр, кДж/моль –394 0 –75 –286
Sо, Дж/(моль∙К)
Решение
а) Найдём вначале энтальпию (теплоту) реакции (по формуле 2.18,а):
ΔНорц = ΔНообр(СН4) + 2 ΔНообр(Н2О) – ΔНообр(СО2) = – 253 кДж/моль. (I.16)
б) Оценим также изменение энтропии в ходе реакции. В условии задачи приведены значения абсолютной энтропии соответствующих веществ. Очевидно, для них справедлива формула, аналогичная (2.18,а):
ΔSорц = Sо(СН4) + 2 Sо(Н2О) – Sо(СО2) – 4 Sо(H2) = – 412 Дж/(моль∙К). (I.17)
в) Тогда, полагая Т = 298 К, для стандартной энергии Гиббса реакции получаем (5.12):
ΔGорц = ΔНорц – TΔSорц = –130 кДж/моль . (I.18)
Значительное отрицательное значение энергии Гиббса свидетельствует о том, что при соответствующих кинетических условиях реакция может самопроизвольно протекать в прямом направлении.
г) Но заметим, что этот итог достигается за счёт лишь одного фактора – энтальпийного (выделения теплоты), тогда как второй фактор (энтропийный) оказывает противоположное (но более слабое) воздействие на общий баланс энергии Гиббса. Т. е. протекание реакции в прямом направлении сопровождается не ростом, а снижением энтропии. Действительно, в ходе реакции 5 молекул двух газов преобразуются лишь в 2 молекулы третьего газа и молекулу жидкости. Т. е. степень свободы (разупорядоченность) системы существенно понижается.
9. В закрытом сосуде идёт обратимая реакция: N2O4 ↔ 2 NO2 .
При t = 25 оC её константа равновесия равна Кр = 0,1 моль/л. В каком направлении идёт реакция при следующих активностях веществ:
а(NO2) = 0,01 моль/л, а(N2O4) = 0,02 моль/л?
Решение
а) «Произведение активностей» равно
Па = а(NO2)2 / а(N2O4) = 0,5 моль/л (I.19)
б) По уравнению изотермы (уравнению Вант-Гоффа) (4.34) определяем энергию Гиббса реакции:
ΔGрц = RT ln (Па/Kp) ≈ –7,4 кДж/моль. (I.20)
в) Т. к. ΔGрц < 0, равновесие реакции сдвинуто вправо.
10. Дана реакция:
NaOH (тв) + СО2 (г) = Na2CO3 (тв) + Н2О (г) .
ΔGообр , кДж/моль –425 –394 –1060 –229
Может ли она протекать в прямом направлении при следующих условиях:
Т = 298 К, Р(СО2) = 10–5 атм, Р(Н2О) = 10–3 атм?
Решение
а) Стандартная энергия Гиббса реакции (4.40,а):
ΔGорц = ΔGообр(Na2CO3) + ΔGообр(Н2О) – ΔGообр(NaOH) – ΔGообр(СО2) = –470 кДж/моль.
. (I.21)
б) Концентрационный член энергии Гиббса реакции (4.33):
RT ln Пс = RT ln [Р(Н2О) / P(СО2)] = RT ln102 = 11,4 кДж /моль. (1.22)
в) Итоговое значение энергии Гиббса реакции при указанных условиях – рассчитывается по уравнению изотермы (4.37):
ΔGрц = ΔGорц + RT ln Пс = –458,6 кДж/моль . (1.23)
Таким образом, прямая реакция является резко экзергонической и будет оставаться таковой при любых физически возможных парциальных давлениях СО2 и Н2О.
11. Пусть вновь (как в задаче 9) в закрытом сосуде идёт обратимая реакция:
N2O4 ↔ 2 NO2 .
При t = 55oC Кр = 1,38∙105 н/м2 (Па). (Заметим: для реакций в газовой среде Кр может выражаться не только в концентрационных единицах, как это было в задаче 9, но и в единицах давления.)
Сколько молей N2O4 надо поместить в сосуд объёмом V = 10л, чтобы после достижения равновесия концентрация продукта реакции установилась на уровне [NO2] = 0,1 моль/л?
Решение
а) Уравнение Клайперона-Менделеева можно записать через концентрацию газа (3.23):
Р = nRT / V = cRT .
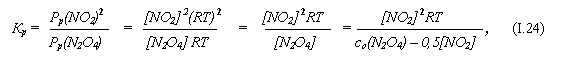 |
б) С учётом этого, обозначая равновесные давления через Рр(Х), а равновесные концентрации – через [X], имеем:
где со(N2O4) – начальная концентрация N2O4, а Т = 328 К.
в) Отсюда
со(N2O4) = 0,5[NO2] + [NO2] 2RT / Кр ≈ 250 моль/м3 = 0,25 моль/л. (1.25)
Заметим: поскольку Кр выражена в н/м2, то концентрации веществ при расчёте должны быть выражены в моль/м3: [NO2] = 0,1 моль/л = 100 моль/м3 .
г) В итоге, искомое начальное количество N2O4 составляет
nо(N2O4) = со(N2O4)∙V ≈ 2,5 моля . (I.26)
12. Дана реакция:
CuSO4∙3H2O (тв) + 2 Н2О (г) ↔ CuSO4∙5H2O (тв) .
Для неё ∆Норц = –106,7 кДж/моль, и при Т1 = 303 К равновесное давление пара воды равно Р1(Н2О) = 1,45 кПа. Найти Кр при Т2 = 293 К.
Решение
а) Константа равновесия при исходной температуре:
Kр,1 = 1 / [Р1(Н2О)]2 ≈ 0,4756 кПа–2 . (I.27)
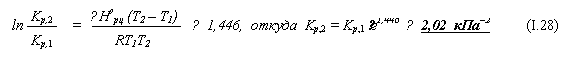 |
б) Значение этой константы при другой температуре можно найти с помощью уравнения изобары химической реакции (5.19):
Как видно, при снижении температуры на 10 градусов константа равновесия рассматриваемой реакции возрастает более чем в 4 раза.
13. В закрытом сосуде идёт обратимая реакция: Н2О + СО ↔ СО2 + Н2 .
Для неё ∆Норц = 41,84 кДж/моль, и при Т1 = 1278 К константа равновесия равна Кр,1 = 1,62. Изначально в сосуд внесено по 1 молю Н2О и СО. Найти количество Н2О и СО в сосуде после достижения равновесия при Т2 = 80.
Решение
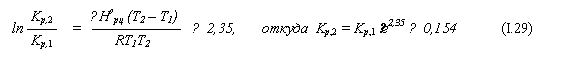 |
а) Как и в предыдущей задаче, по уравнению изобары химической реакции находим константу равновесия при второй температуре:
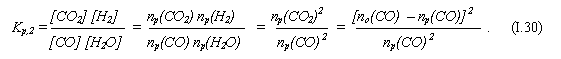 |
б) С другой стороны, эта константа, по определению, равна
Здесь nр(Х) = [X]∙V – количество (в молях) компонента Х в сосуде после достижения системой равновесного состояния.
Т. к. реагенты введены в систему в одинаковых количествах (no(СО) = nо(H2O) = 1 моль), то, исходя из стехиометрии реакции, величины nр(Х) попарно равны (для реагентов и для продуктов):
nр(СО) = nр(H2O), nр(СО2) = nр(H2) .
Причём, появляющееся в системе количество каждого из продуктов равно убыли количества каждого из реагентов; в частности
nр(СО2) = no(СО) – nр(СО).
Всё это учтено в преобразованиях (I.30).
в) Выражая из конечного соотношения np(СО), получаем искомое содержание СО в сосуде при равновесном состоянии системы:
nр(СО) = no(СО) / (√Kp + 1) ≈ 0,718 моля . (I.31)
Таково же и равновесное содержание воды.
РАЗДЕЛ 2.
ФАЗОВЫЕ РАВНОВЕСИЯ И
РАСТВОРЫ НЕЭЛЕКТРОЛИТОВ
Глава 6. ОБЩИЕ ЗАКОНОМЕРНОСТИ ФАЗОВЫХ ПЕРЕХОДОВ
6.1. Фазы системы
Ключевое понятие в данном разделе — фаза.
Фаза — это совокупность гомогенных частей системы, имеющих одинаковые химические, физические и термодинамические характеристики во всех своих точках. Поясним это определение.
1. Прежде всего, для образования разных фаз достаточно присутствия в системе разных агрегатных состояний одного и того же вещества.
а) Известны три агрегатных состояния вещества — газообразное, жидкое и твердое. Они отличаются по своим физическим (плотность, теплоемкость) и термодинамическим (ΔНообр, ΔSообр и т. д.) параметрам. А это, в соответствии с приведенным определением, и означает образование разных фаз.
б) I. Пример однокомпонентной двухфазной системы — кусочки льда в воде. Здесь — две фазы одного вещества: жидкая и твердая.
II. Включение в систему водяного пара над поверхностью позволяет получить трехфазную однокомпонентную систему, куда входит еще и газообразная фаза.
в) Переход же вещества из одного агрегатного состояния в другое называется, как известно, фазовым превращением или фазовым переходом. Последний из этих терминов уже встречался в п. 3.4.
Итак, одно и то же вещество может образовывать несколько фаз в системе.
2. С другой стороны, одну и ту же фазу могут образовывать несколько разных веществ. Так обстоит дело тогда, когда вещества взаимно растворены друг
в друге (например, если это — хорошо смешивающиеся жидкости), или речь
идет о растворе каких-то веществ в некотором растворителе.
Таким образом, истинный раствор — это однофазная система.
3. В третьих случаях разные вещества образуют в системе разные фазы.
а) Пример — смесь воды и масла. Даже если ее хорошо взболтать, она быстро
расслаивается на две жидкие фазы — фазу масла и фазу воды. Последние
различаются не только по физическим и термодинамическим, но и по хими-
ческим характеристикам.
б) Если добавить в эту систему и третий компонент —
например, раздробленный уголь, образуется третья фаза — твердая. В данном
примере число фаз совпадает с числом компонентов.
4. а) Но в общем случае, как следует из предыдущих примеров, количество фаз в системе может не совпадать с количеством компонентов. Число фаз обо-
значается буквой Ф. Системы, состоящие из одной фазы (Ф = 1), называются
гомогенными, а из нескольких фаз (Ф ≥ 2) — гетерогенными.
б) Заметим также, что фаза может быть непрерывной, а может быть дисперс-
ной, т. е. состоять из отдельных фрагментов или частиц. Примеры дисперсных
фаз — глыбки льда; капельки масла в воде; пузырьки воздуха, появляющиеся
в воде при нагревании; частицы измельченного угля; пыль в воздухе и т. д.
В каждом из этих примеров дисперсная фаза распределена в какой-либо
второй — непрерывной — фазе.
6.2. Число независимых компонентов
Другим важным параметром системы (наряду с параметром Ф) является
число независимых компонентов — К. Слово «независимых» часто опускают
и говорят просто: «число компонентов».

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 |
