


Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
1. В итоге, с законом Рауля связаны 4 явления, касающиеся влияния растворенного вещества на свойства раствора. Это
a) понижение давления насыщенного пара растворителя: ΔР1 /P01 = Х2 , (8.17, в)
б) понижение температуры замерзания раствора: ΔТк = Kэб b2 , (8.20)
в) повышение температуры кипения: ΔTз = – Кз b2, (8.21)
г) наличие осмотического давления: П ≈ с2RT. (8.28)
2. Все эти явления объединяют в группу так называемых коллигативных свойств растворов.
а) Термин означает, что выраженность данных свойств зависит только от
количества частиц растворенного вещества в единице объема и не зависит от
их природы.
б) Однако такое утверждение (равно как и выписанные соотношения) спра-
ведливо только для идеальных растворов. В случае же неидеальных растворов
имеет значение и природа растворенного вещества.
8.10. Активности веществ
В вышеприведенных выражениях для коллигативных свойств растворов
фигурируют концентрации растворенных веществ. Использовались концентра-
ции и тогда, когда записывались формулы для константы равновесия, энергии
Гиббса химической реакции, химического потенциала, например:
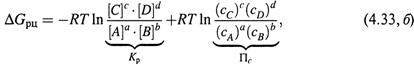 |
1. а) Но на самом деле, как уже отмечалось для химического потенциала (6.9, б), во всех этих формулах следует писать не просто концентрации, а так называемые эффективные концентрации, или активности веществ. Последние связаны с обычными концентрациями с помощью коэффициентов активности:
причем индексы (X, с, b) в обозначениях активности обычно не пишут. Так что под активностью иногда понимают эффективную молярную долю, в других случаях — эффективную молярную концентрацию, а в третьих — эффективную моляльную концентрацию.
б) С учетом сделанного замечания дадим определение. Активность вещества в растворе — это концентрация идеального раствора, которая требуется для проявления точно таких же коллигативных, химических и прочих свойств, как у данного реального раствора.
в) Следовательно, чтобы на практике определить коэффициент активности, необходимо измерить какое-либо свойство раствора, по соответствующей формуле рассчитать эффективную концентрацию и сравнить последнюю с истинной концентрацией.
2. Пример. Пусть для раствора вещества с c2 = 0,1 М измерили осмотическое давление и по формуле Вант-Гоффа нашли, что такое же давление создает идеальный раствор с концентрацией

Тогда коэффициент активности равен
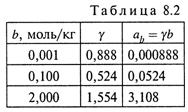 3. При очень низких концентрациях вещества коэффициент активности близок к 1,0 (рис.8.6); при повышении концентрации он снижается (становясь много меньше 1,0); при еще большем увеличении концентрации начинает повышаться и порой даже становится больше 1,0. Некоторые значения коэффициента активности для СаСl2 приведены в табл. 8.2.
3. При очень низких концентрациях вещества коэффициент активности близок к 1,0 (рис.8.6); при повышении концентрации он снижается (становясь много меньше 1,0); при еще большем увеличении концентрации начинает повышаться и порой даже становится больше 1,0. Некоторые значения коэффициента активности для СаСl2 приведены в табл. 8.2.
4. Почему активность обычно отличается от концентрации?
Все дело во взаимодействии частиц вещества друг с другом. Например, ионы K+ и Cl–, на которые распадается в растворе KCl, создают друг вокруг друга многочисленные ионные оболочки. Поэтому некоторая доля частиц как бы выключается из взаимодействия с растворителем и другими компонентами раствора.
5. Итак, в выражениях, где до сих пор писались концентрации, на самом деле
надо писать активности, например:
μi = μ0i + RT ln (γi Xi) , (8.35,а)
(aC)c (aD)d
ΔGрц = ΔG0рц + RT ln –––––––––– = ΔG0рц + RT ln Па . (8.35,б)
(aA)a (aB)b
8.11. Уравнение Гиббса-Дюгема
1. В физической химии известно так называемое уравнение Гиббса-Дюгема.
Оно имеет весьма общий смысл, но с его помощью, в частности, можно
связать коэффициенты активности компонентов раствора. Вывод уравнения таков.
а) Полная энергия Гиббса системы выражается через химические потенциалы компонентов:
откуда
б) С другой стороны, известно выражение:
в) Сопоставление дает:
или, при постоянных давлении и температуре,
2. а) Это и есть уравнение Гиббса-Дюгема, записанное для химических потенциалов. В данной записи оно означает, что для компонентов раствора изменения химических потенциалов взаимосвязаны. Например, увеличение потенциала одного компонента может происходить только за счет уменьшения потенциала другого компонента.
б) Так, увеличение концентрации растворенного вещества повышает его химический потенциал. Но в результате снижается молярная доля растворителя — значит, уменьшается его химический потенциал.
3. а) Подставим в (8.37, б) формулу (8.35, а) и поделим все на ∑ ni:
б) Для двухкомпонентной системы нетрудно убедиться, что второе слагаемое
уравнения (8.38) равно нулю:
в) Поэтому первый член уравнения (8.38) тоже равен нулю, и это для двукомпонентной системы можно записать так:
4. Вывод таков. Если с ростом X2 уменьшается коэффициент активности растворяемого вещества  , то одновременно увеличивается коэффициент активности растворителя . И наоборот. Зная, как уменьшается γ2, можно рассчитать и увеличение γ1.
, то одновременно увеличивается коэффициент активности растворителя . И наоборот. Зная, как уменьшается γ2, можно рассчитать и увеличение γ1.
8.12. Коллигативные свойства растворов электролитов
В данном разделе (т. е. в разделе 2), как следует из его названия (см. стр. 61), рассматриваются растворы неэлектролитов. Однако чтобы не возвращаться еще раз к коллигативным свойствам, обратимся в этом пункте к растворам электролитов.
1. В случае электролитов в формулах для коллигативных свойств (п. 8.9)
поправку на особенности реальных растворов делают не путем замены концентраций на активности, а путем введения изотонического коэффициента i:
2. Для сильных электролитов этот коэффициент равен
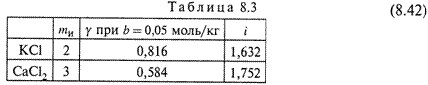 |
где γ — коэффициент активности, а mи — количество ионов, на которое
распадается электролит. Два примера расчета изотонического коэффициента
по формуле (8.42) приведены в табл. 8.3.
3. а) В случае же слабых электролитов отличие ί от mи обусловлено, главным образом, неполной диссоциацией (а не взаимодействием диссоциирующих
частиц). Поэтому коэффициент активности можно считать равным 1, и вся
задача состоит в расчете среднего числа частиц, образующихся из одной молекулы.
б) Используя степень диссоциации α, можно записать:
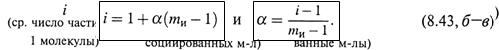 |
откуда
в) Последняя формула используется тогда, когда изотонический коэффициент
определен экспериментально (например, по изменению температуры замерзания раствора, т. е. по формуле (8.41,в)). В этом случае, как видно, можно вычислить степень диссоциации слабого электролита.
8.13. Растворение газов в жидкости (законы Генри и Сеченова)
Опять обратимся к растворам неэлектролитов. Некоторые особенности
имеет растворение газов.
1. а) В отличие от твердых тел, здесь растворяемое вещество тоже (как и растворитель) является летучим, поэтому можно рассматривать фазовое равновесие в системе раствор — газ над раствором.
б) Для идеального раствора к этому равновесию тоже применим закон Рауля (8.12), который в данном случае следует записать так:
Следовательно, давление газа над раствором (как и любого летучего компонента раствора) пропорционально молярной доле газа в растворе. Здесь
в) Однако для реальных систем вместо
2. а) Нередко это соотношение рассматривают в обратную сторону:
б) Перейдем к молярной концентрации:
Тогда получаем:
где Kp— константа растворимости, которую можно представить в виде:
в) Тем самым выведен закон Генри: концентрация растворенного в жидкости газа пропорциональна парциальному давлению этого газа над раствором.
3. Уточним природу константы растворимости. (Сейчас она определена лишь
через другую эмпирическую константу — Kг.)
а) Запишем условие равенства химических потенциалов газа в растворе и над ним:
в котором ![]() — химический потенциал растворенного газа при его единичной молярной концентрации, а
— химический потенциал растворенного газа при его единичной молярной концентрации, а ![]() — потенциал газа над раствором при его единичном парциальном давлении.
— потенциал газа над раствором при его единичном парциальном давлении.
б) Обозначим:
Тогда находим:
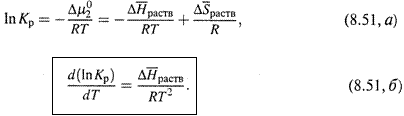 |
в) Отсюда следует температурная зависимость Kp— такая же, как для константы равновесия химической реакции (п. 5.9):
г) Если ![]() (растворение газа — экзотермический процесс), то с ростом температуры Kp уменьшается, и растворимость газа снижается.
(растворение газа — экзотермический процесс), то с ростом температуры Kp уменьшается, и растворимость газа снижается.
Данное явление весьма характерно для газов.
4. На растворимость газов влияют и соли, присутствующие в растворе. Это
устанавливается эмпирическим законом Сеченова:
С ростом концентрации солей растворимость газа тоже понижается.
8.14. Растворимость твердых веществ в жидкости
А как зависит от температуры растворимость твердого вещества? Подход
и принципиальные результаты здесь совершенно таковы же, что и для газов.
а) Воспользуемся формальным представлением растворения в виде химической реакции:
(Вещество 2)тв ↔ (Вещество 2)р-р (8.10,а)
б) Как известно, константа равновесия в данном случае просто совпадает
с равновесной концентрацией растворяемого вещества:
Кр = [Вещество 2] . (8.10,б)
в) Для этой константы-концентрации уравнение изобары (5.20) записывается так:
г) Вытекающий из него вывод аналогичен таковому для растворимости газов:
если
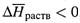 , то при росте температуры растворимость вещества понижается. Однако в случае твердых веществ чаще наблюдается обратная ситуация:
, то при росте температуры растворимость вещества понижается. Однако в случае твердых веществ чаще наблюдается обратная ситуация:
Поэтому их растворимость при нагревании обычно увеличивается.
Краткое содержание главы 8
Глава посвящена частному виду двухкомпонентных систем – РАСТВОРАМ.
1. Для их характеристики наиболее часто используются молярные (сi) и моляльные (bi) концентрации, а также молярная доля (Хi), связанные следующими соотношениями:
сi ≈ Xi ni /Vo , Xi ≈ bi Mi
2. В т. н. ИДЕАЛЬНЫХ РАСТВОРАХ все межмолекулярные взаимодействия одинаковы по характеру и силе. Для таких растворов изменение энергии Гиббса составляет:
а) при образовании газового раствора –
ΔGо = (n1 + n2) RT [X1 ln X1 + X2 ln X2] ,
б) в жидких растворах – при повышении концентрации растворённого вещества с X2 до X2′:
ΔGо = n1 RT ln ( X1′ /X1 ) + n2 RT ln ( X2′ /X2 ) + Δn2 RT ln X2′ .
 3. Затем речь шла о ФАЗОВОМ РАВНОВЕСИИ МЕЖДУ РАСТВОРИТЕЛЕМ И ЕГО ПАРОМ над раствором. Был сформулирован ЗАКОН РАУЛЯ: давление насыщенного пара над ИДЕАЛЬНЫМ раствором пропорционально молярной доле растворителя:
3. Затем речь шла о ФАЗОВОМ РАВНОВЕСИИ МЕЖДУ РАСТВОРИТЕЛЕМ И ЕГО ПАРОМ над раствором. Был сформулирован ЗАКОН РАУЛЯ: давление насыщенного пара над ИДЕАЛЬНЫМ раствором пропорционально молярной доле растворителя:
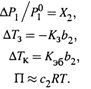 4. Отсюда вытекают КОЛЛИГАТИВНЫЕ ЭФФЕКТЫ. Растворение вещества в растворителе приводит к:
4. Отсюда вытекают КОЛЛИГАТИВНЫЕ ЭФФЕКТЫ. Растворение вещества в растворителе приводит к:
а) понижению давления насыщенного пара растворителя,
б) понижению температуры замерзания раствора,
в) повышению температуры кипения,
г) возникновению осмотического давления,
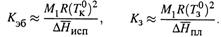 При этом эбулиоскопическая и криоскопическая константы определяются природой только растворителя:
При этом эбулиоскопическая и криоскопическая константы определяются природой только растворителя:
5. Для растворов ЭЛЕКТРОЛИТОВ вводится поправочный множитель — ИЗОТО-НИЧЕСКИЙ КОЭФФИЦИЕНТ i :
![]()
причем для сильных и для слабых электролитов он вычисляется так:
6. а) И в общем случае эффективные концентрации (АКТИВНОСТИ) часто отличаются от истинных:
аX = γХ, аc = γс, ab = γb .
б) КОЭФФИЦИЕНТЫ АКТИВНОСТИ γ зависят от концентрации веществ, что выражается УРАВНЕНИЕМ ГИББСА-ДЮГЕМА:
(1 – Х2) d (ln γ1) = – X2 d (ln γ2) .
7. РАСТВОРИМОСТЬ ГАЗА зависит от его парциального давления над раствором (ЗАКОН ГЕНРИ) и концентрации солей в растворе (ЗАКОН СЕЧЕНОВА):
с2 ≈ КР Р2 , ln KР = ln KР0 – k cсоль .
Глава 9. ЖИДКИЕ СМЕСИ С НЕОГРАНИЧЕННОЙ
РАСТВОРИМОСТЬЮ КОМПОНЕНТОВ
Теперь рассмотрим растворы жидкости в жидкости, которые правильней называть смесями. Смеси различаются по взаимной растворимости жидкостей, что связано с природой последних и характером межмолекулярного взаимодействия.
Рассмотрим вначале простейший тип смесей. Как обычно, будем иметь в виду систему под поршнем.
9.1. Смеси, подчиняющиеся закону Рауля (идеальные смеси): давление и состав пара
1. Исходные условия. а) Будем также считать, что жидкости
I. неограниченно растворимы друг в друге и
II. обе являются летучими, т. е. дают над раствором пар.
б) Введём обозначения: 1 — менее летучая жидкость, 2 — более летучая, т. е. у первой жидкости — ниже давление насыщенного пара над чистым веществом и, соответственно, выше температура кипения:
в) Вспомним также условие идеальности раствора, согласно которому все виды межмолекулярных взаимодействий в системе — 1–1, 2–2 и 1–2 — одинаковы
по характеру и силе. Примерами являются смеси близких по природе жидкостей: бензол–толуол, гексан–гептан, метанол–этанол.
2. а) Для идеальной смеси закон Рауля (8.12) применим к обоим летучим компонентам:
откуда
![]()
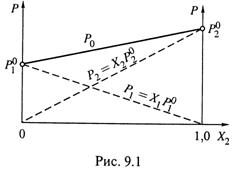 б) Получается, что общее давление пара над смесью линейно зависит от состава смеси. Это изображено на рис. 9.1, где
б) Получается, что общее давление пара над смесью линейно зависит от состава смеси. Это изображено на рис. 9.1, где
- по оси абсцисс отложена молярная доля в жидкой фазе более летучего компонента,
- пунктирными линиями показаны парциальные давления паров каждого из двух
компонентов (при разном составе смеси)
- и сплошной линией — общее давление
пара над раствором.
3. а) Нетрудно определить состав пара при том или ином составе жидкой фазы. Молярные доли каждого вещества в паре обозначим штрихом. Тогда, например, доля в паре второго компонента такова:
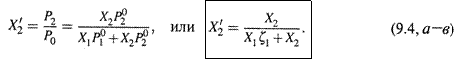 |
 Последнее выражение получено путем деления числителя и знаменателя на
Последнее выражение получено путем деления числителя и знаменателя на ![]() , а ζ1— величина, меньшая единицы:
, а ζ1— величина, меньшая единицы:
![]()
поскольку, в соответствии с условием, первый компонент является менее летучим.
б) Заметим: так как X1 + X2 = 1, то знаменатель выражения (9.4, в) меньше единицы:
в) Таким образом, доля второго (более летучего) компонента в паре выше, чем в жидкой смеси. В этом заключается первый закон Коновалова: по сравнению с жидкой фазой, состав газовой фазы обогащен более летучим компонентом.
На рис. 9.2 это выражается в том, что кривая зависимости ![]() от X2 идет выше диагонали единичного квадрата. График же данной зависимости нетрудно построить, используя формулу (9.4, в).
от X2 идет выше диагонали единичного квадрата. График же данной зависимости нетрудно построить, используя формулу (9.4, в).
9.2. Идеальные смеси:
полный вариант диаграммы давления
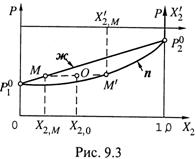 1. а) Нередко на диаграмме давления (рис. 9.3), кроме зависимости P0 от состава жидкости X2 (линии ж), изображают также линию, связывающую P0
1. а) Нередко на диаграмме давления (рис. 9.3), кроме зависимости P0 от состава жидкости X2 (линии ж), изображают также линию, связывающую P0
с составом газовой фазы ![]() (линия п). Для этого удобно ввести еще одну
(линия п). Для этого удобно ввести еще одну
координатную ось — ![]() (хотя часто обходятся и без неё).
(хотя часто обходятся и без неё).
б) Тогда построение таково (см. рис. 9.3). Для произвольного значения X2,М по формулам (9.4) рассчитывают соответствующее значение ![]() (большее X2,М) и находят точку пересечения этой ординаты
(большее X2,М) и находят точку пересечения этой ординаты ![]() с изобарой, проходящей через точку М. Тем самым получают точку М', принадлежащую линии п.
с изобарой, проходящей через точку М. Тем самым получают точку М', принадлежащую линии п.
2. Линиями ж и п диаграмма разделяется на три области.
а) Над линией ж система имеет только жидкую фазу (имеется в виду, что система находится под поршнем). В любой точке этой области внешнее давление (создаваемое поршнем) больше возможного давления пара смеси, так что пар не образуется.
б) Между линиями возможно существование двух фаз: и жидкой, и газообразной. Например, в точке О (состав — X2,0, внешнее давление — P0 = Pм) сосуществуют жидкая фаза с составом X2,М и газовая с составом ![]() .
.
в) А под линией n система имеет только одну газовую фазу.
3. Отметим также, что состав газовой фазы зависит от температуры {закон
Вревского). Это следует из формулы (9.4,в). В ней фигурирует ζ1 — отношение
величин ![]() и
и ![]() . Если эти величины с ростом температуры изменяются
. Если эти величины с ростом температуры изменяются
непропорционально, то ![]() оказывается функцией температуры.
оказывается функцией температуры.
9.3. Идеальные смеси: диаграммы кипения
1. Теперь обратимся к температуре кипения смеси.
а) Если компоненты смеси различаются по своей летучести (значениям ![]() и
и ![]() ), то они различаются и по температурам кипения в чистом состоянии.
), то они различаются и по температурам кипения в чистом состоянии.
б) У менее летучей жидкости 1, очевидно, данная температура выше:
Это показано на рис. 9.4 соответствующими точками на осях температуры.
2. Для смеси же температура кипения, как тоже очевидно, будет зависеть от её состава.
а) Понятно, что с ростом X2 (увеличением доли более летучего компонента) Тк смеси будет уменьшаться — от Тк,1 до Тк,2 (нижняя кривая на рис. 9.4).
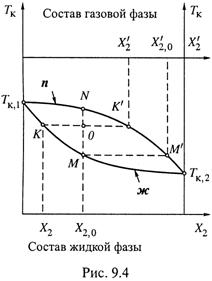 б) В отличие от зависимости P0(X2) ((9.3) и рис. 9.1), рассматриваемая сейчас зависимость является нелинейной.
б) В отличие от зависимости P0(X2) ((9.3) и рис. 9.1), рассматриваемая сейчас зависимость является нелинейной.
Действительно, количественную зависимость Тк от X2 можно получить, если исходить из условия: общее давление пара над смесью равно внешнему давлению:
где для величин
Без дальнейших вычислений понятно, что
зависимость Тк от Х2 окажется нелинейной.
в) Верхняя кривая (п) связывает температуру кипения и состав газовой фазы. Построение ее происходит так же, как построение линии п на рис. 9.3; так что в итоге точке М линии ж соответствует точка М' линии п.
3. а) Диаграмма опять-таки разделяется на три области, но в отличие от диаграммы давления порядок расположения областей обратен: область пара — над линией п, а область жидкой фазы — под линией ж.
б) Кроме того, диаграмма позволяет прогнозировать, как будут изменяться состав и масса каждой фазы в процессе кипения смеси.
4. Состав фаз. Пусть исходный состав жидкости (находящейся в системе под поршнем) — X2,0.
а) По достижении температуры кипения (точки М на нижней кривой диаграммы рис. 9.4) в системе начинает образовываться пар с составом ![]() (точка М' на верхней кривой).
(точка М' на верхней кривой).
б) В процессе кипения переход в пар второго (более летучего) вещества происходит легче, чем первого. Поэтому содержание второго компонента в жидкой фазе (величина X2) неуклонно снижается. Движение идет по нижней кривой влево от точки М.
в) Соответственно, снижается содержание этого вещества и в паре. Движение по верхней кривой от точки М' — тоже влево.
г) Таким образом, на первых стадиях процесса преимущественно испарялся компонент 2 и пар был им обогащен. Со временем же постепенно возрастают:
- доля компонента 1 в жидкой фазе,
- температура кипения этой фазы,
- доля компонента 1 в паре.
д) Напомним: предполагается, что пар никуда из системы не отводится. Тогда суммарный состав системы (с учетом и жидкости, и пара) остается постоянным: он численно равен X2,0 и на рис.9.4 отражается одной из точек вертикали МN.
е) Ясно также, что при полном испарении обоих компонентов состав пара будет описываться точкой N, т. е. сравняется с составом исходной жидкой смеси.
5. Масса фаз. Кроме состава фаз смеси на разных стадиях испарения, можно оценить и массы фаз.
а) Допустим, на некоторой стадии кипения состав жидкой фазы отражается
точкой К, а парообразной — точкой К', тогда суммарное состояние системы
отражается точкой О (местом пересечения горизонтали КК' с вертикалью МN).
б) Можно убедиться в перекрёстном правиле рычага:
- отрезок ОК на рис. 9.4, примыкающий к нижней кривой (ж), пропорционален массе газовой фазы,
- а отрезок ОК', примыкающий к верхней кривой (п), — массе жидкой фазы.
в) Действительно, в состоянии О молярная доля вещества 2 в системе
(сохраняющая исходное значение) может быть вычислена так:
где n0 — общее число молей обоих компонентов в обеих фазах, а nж и nг —
количество молей обоих компонентов, соответственно, в жидкой и газовой фазах.
Подставляя n0 = nж + nг, после простых преобразований получаем:
что и означает правило рычага.
9.4. Смеси, отклоняющиеся от закона Рауля
1. а) Итак, для идеальной смеси двух жидкостей общее давление пара линейно зависит от молярной доли компонентов:
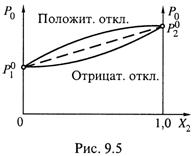
но для реальных растворов зависимость часто отклоняется от линейной.
б) На диаграмме рис. 9.5 показано: могут быть как положительные отклонения, когда общее давление пара P0 больше того, что ожидается по формуле (9.3)), так и отрицательные отклонения (P0 меньше ожидаемого значения).
 |
в) Эти ситуации суммированы в табл. 9.1.
2. В частности, для смесей с положительным отклонением из данной таблицы видно следующее.
а) В этих смесях энергия взаимодействия между молекулами разных компонентов (E12) меньше, чем, в среднем, между одинаковыми молекулами. Следовательно, при образовании таких смесей энергия межмолекулярных взаимодействий, приходящаяся на одну частицу, уменьшается и молекулы становятся более подвижными.
б) В силу этого образование смесей идет с поглощением теплоты (при условии постоянства температуры):
в) Испаряются молекулы из смеси в таком случае легче, чем из чистых жидкостей: теплота испарения снижается —
г) В результате парциальное давление пара компонента ![]() выше, чем в случае идеальной смеси
выше, чем в случае идеальной смеси ![]() .
.
3. а) Можно дать еще одну интерпретацию положительного отклонения. В п. 8.5 выведен закон Рауля для идеальных растворов. В случае неидеальных раство-
ров или смесей можно повторить тот же вывод, но коэффициент распреде-
ления вещества между фазами записать не через концентрации, как в (8.14),
а через активности:
б) Тогда, вместо
 , мы придем к выражению
, мы придем к выражению
где величина
в) Из (9.12,а) и вытекает объяснение положительного отклонения через активности:
Следовательно, данный тип отклонения наблюдается тогда, когда коэффи-
циенты активности компонентов в жидкой фазе выше, чем в газовой фазе.
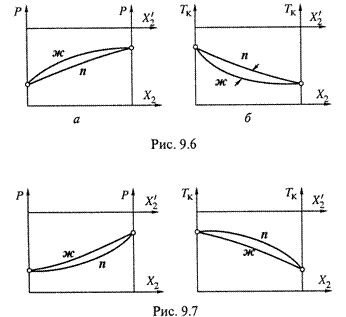 4. а) Обе диаграммы давления и кипения при положительном отклонении показаны на рис. 9.6,а–б. В частности, температура кипения смеси при любом её составе (кроме крайних случаев) оказывается ниже, чем для идеальной смеси. Поэтому обе кривые на диаграмме рис. 9.6,б (ж, зависимость Tк от X2, и n, связь
4. а) Обе диаграммы давления и кипения при положительном отклонении показаны на рис. 9.6,а–б. В частности, температура кипения смеси при любом её составе (кроме крайних случаев) оказывается ниже, чем для идеальной смеси. Поэтому обе кривые на диаграмме рис. 9.6,б (ж, зависимость Tк от X2, и n, связь ![]() с Tк) как бы прогнуты вниз.
с Tк) как бы прогнуты вниз.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 |
