


Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
б) Тогда создаваемая ЭДС просто равна искомому потенциалу, если пара Oxι/Rdι играет в реакции с водородной парой роль окислителя:
в) Если же, наоборот, данная пара выступает в качестве восстановителя, её
потенциал равен измеренной ЭДС, взятой с обратным знаком:
4. а) Электродные потенциалы, измеренные относительно стандартного водородного полуэлемента, называются редокс-потенциалами. Поскольку другой
шкалой измерения обычно не пользуются, то понятия «электродные потенциалы» и «редокс-потенциалы» часто совпадают.
б) Итак, редокс-потенциалы характеризуют окислительно-восстановительные
свойства соответствующей пары по сравнению со свойствами пары H+/H2
(в стандартных условиях).
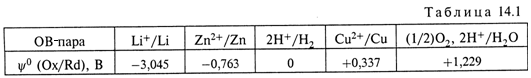 |
в) Стандартные редокс-потенциалы некоторых окислительно-восстановительных пар приведены в табл. 14.1.
5. а) При отрицательных значениях редокс-потенциала сродство ОВ-пары к электронам меньше, чем у водородной пары. Действительно, это соответствует
уравнению (14.30), в котором пара Oxι/Rdι выступает в качестве восстановителя, т. е. отдает электроны водородной паре.
б) При положительных же значениях Ψ0(Oxι/Rdι), напротив, сродство ОВ-пары к электронам выше, чем у пары H+/H2.
14.11. Примеры расчета ЭДС для элемента Даниэля-Якоби
Продемонстрируем использование полученных формул на примере элемента Даниэля–Якоби.
1. Стандартные условия.
а) Применим формулу (14.10), учитывая (14.15) и табл. 14.1:
Очевидно, таким же будет значение ЭДС при любых, но равных, активностях ионов Cu2+ и Zn2+ в полуэлементах.
б) Зная же ![]() , по формуле (14.17,б) нетрудно найти стандартную энергию Гиббса реакции:
, по формуле (14.17,б) нетрудно найти стандартную энергию Гиббса реакции:
Результат совпадает с полученной иным способом оценкой (14.4,б).
2. Теперь пусть а(Cu2+) = 0,01 М, а(Zn2+) =0,1 М.
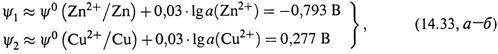 |
а) Здесь для каждого потенциала следует применить уравнение Нернста (Петерса). Используем его адаптированный вариант (14.26):
б) Можно рассчитать ЭДС и минуя предварительную оценку потенциалов. Для этого достаточно использовать уравнение Нернста, относящееся непосредственно к ЭДС (14.17,а):
По такому же принципу проводятся расчеты и во многих других случаях.
14.12. Кажущиеся потенциалы
1. а) Если в соответствующей ОВР участвуют протоны (в качестве реагентов
или продуктов), то, как было отмечено для водородного электрода (14.28,б),
стандартное значение ЭДС или отдельного потенциала относится к рН = 0.
б) Более же интересны для биолога так называемые кажущиеся потенциалы — Ψi', которые относятся к рН = 7,0 (и единичным активностям всех прочих,
кроме протонов, реагентов).
2. а) Оценить эти значения можно, вновь исходя из стандартных значений и снова используя уравнение Нернста, – но на этот раз вариант (14.23,б) данного уравнения (с учётом формулы (14.24)).
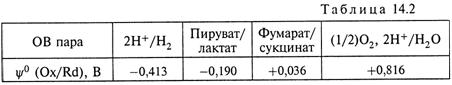 |
б) Приведем кажущиеся редокс-потенциалы некоторых ОВ пар (табл. 14.2).
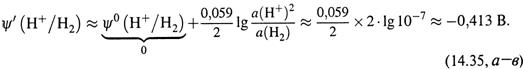 |
в) Например, для водородного полуэлемента получаем:
г) Если обратиться к последней ОВ-паре табл.14.2, то нетрудно видеть, что для неё кажущееся значение потенциала меньше стандартного (1,223 В; см. табл. 14.1) на те же 0,413 В, что и для водородной пары.
Расчеты производятся так же, как и в случае стандартных потенциалов.
14.13. Механизм возникновения электродных потенциалов
1. Теперь попробуем более детально представить, как возникают электродные потенциалы. Допустим, металлическая пластинка опущена в воду (рис. 14.5).
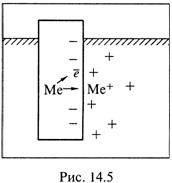 а) В чистых металлах валентные электроны располагаются достаточно свободно, и в узлах кристаллической решетки находятся, по существу, ионы.
а) В чистых металлах валентные электроны располагаются достаточно свободно, и в узлах кристаллической решетки находятся, по существу, ионы.
Поэтому под действием растворителя (воды) некоторое количество ионов диффундирует в жидкую фазу, и пластинка приобретает отрицательный заряд, препятствующий дальнейшему вымыванию ионов. При некоторой величине этого заряда устанавливается динамическое равновесие.
б) У поверхности же пластинки концентрируются (но располагаются относительно диффузно) ионы — того же металла или иные, если таковые есть в растворе. В результате формируется так называемый двойной электрический слой.
А между самой пластинкой и жидкой фазой возникает разность потенциалов, называемая электродной — ∆Ψэ.
 2. а) Условно можно принять, что на самой пластинке потенциал равен ∆Ψэ, а на достаточно большом удалении от неё — нулю.
2. а) Условно можно принять, что на самой пластинке потенциал равен ∆Ψэ, а на достаточно большом удалении от неё — нулю.
б) Из-за диффузного расположения ионов потенциал среды по мере удаления от пластинки убывает нелинейно, а разность потенциалов между пластинкой и точкой среды тоже нелинейно возрастает от 0 до ∆Ψэ (рис. 14.6).
3. Добавим в раствор соль того же металла.
а) Очевидно, выход из пластинки ионов Me+ затрудняется — и тем сильнее, чем выше концентрация ионов в растворе. В зависимости от этой концентрации, свойств металла и растворителя, результирующий заряд пластинки может оставаться отрицательным, а может и вообще менять знак на обратный.
б) В последнем случае преобладает включение ионов в состав пластинки. Соответственно, меняется также ∆Ψэ..
4. а) Наконец, представим, что мы соединяем проводником две разные металлические пластинки, опущенные в растворы своих солей. Концентрация электронов в пластинках оказывается различной, что приводит к их перетеканию в ту или иную сторону. Это нарушает равновесие у поверхности обеих пластинок.
б) На пластинке, отдающей электроны, облегчается выход в раствор новых
порций ионов Me+, а на пластинке, получающей электроны, облегчается осаждение ионов металла. Тем самым во внешней цепи поддерживается постоянный ток.
в) Но если речь идет об электродах, которые в ходе реакции не
изменяются (как на рис. 14.3), тогда в основе возникновения ∆Ψэ лежит обмен
не ионами, а электронами, совершаемый между металлом и парой Ox/Rd в растворе.
14.14. Составные части ЭДС гальванического элемента
В завершение этой главы уточним, из чего складывается измеряемая ЭДС
элемента.
1. а) Согласно закону Кирхгофа, суммарные падения напряжения (разность потенциалов) во внешней и во внутренней цепи элемента одинаковы по величине и
противоположны по знаку:
б) Следовательно, речь идет об определении всех слагаемых правой части
этого равенства. Или, как говорят, скачков потенциала во внутренней цепи эле-
мента. Такие скачки создаются на каждой границе фаз в элементе.
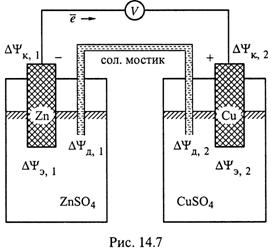 2. а) Два из них, причём самые важные, нам уже известны: это рассмотренные в предыдущем пункте электродные разности потенциалов в каждом из двух полуэлементов, ΔΨэ,1 и ΔΨэ,2 (рис.14.7). Они возникают на границах металлических электродов с жидкими фазами полуэлементов.
2. а) Два из них, причём самые важные, нам уже известны: это рассмотренные в предыдущем пункте электродные разности потенциалов в каждом из двух полуэлементов, ΔΨэ,1 и ΔΨэ,2 (рис.14.7). Они возникают на границах металлических электродов с жидкими фазами полуэлементов.
б) Заметим: записывая для представленного элемента любую разность потенциалов ΔΨi, следует располагать потенциалы фаз в порядке, противоположном ходу часовой стрелки. С учётом этого,
3. а) Еще две границы раздела — между растворами электролитов. В нашем
случае это границы раствора KCl в солевом мостике с раствором ZnSO4 в одном полуэлементе и с раствором CuSO4 — в другом.
б) Соответственно, говорят о диффузионных разностях потенциалов, ΔΨд,1 и ΔΨд,2, возникающих потому, что ионы (компоненты растворов) отличаются по концентрации (I) и подвижности (II).
I. Так, ионы того или иного вида диффундируют через границу раздела в раствор с меньшей концентрацией именно этих ионов. Например, ионы Zn2+и ![]() — в раствор солевого мостика, а ионы K+ и Cl - — в раствор CuSO4.
— в раствор солевого мостика, а ионы K+ и Cl - — в раствор CuSO4.
II. А из-за различия в подвижности эти перемещения происходят с разной скоростью, что и приводит к появлению ΔΨд,i.
в) Рассмотрим сумму величин ΔΨд,1 и ΔΨд,2:
Здесь ΨXY,1 и ΨXY,2 — потенциалы растворa KCl соответственно на границе с первым и вторым полуэлементами.
В силу высокой подвижности ионов К+ и Cl– эти потенциалы можно считать практически одинаковыми (хотя не абсолютно: иначе ионы К+ и Cl– вообще бы не перемещались). Тогда, принимая, что ΨXY,1 ≈ ΨXY,2, имеем:
min (ΔΨд,1 + ΔΨд,2 ) = ΨZnSO4 – ΨCuSO4 . (14.38,б)
г) Таким образом, выбор в качестве солевого мостика насыщенного раствора KCl вовсе не устраняет диффузионную разность потенциалов, а только сводит её к минимально возможному значению, которая определяется природой полуэлементов.
4. Наконец, контактные разности потенциалов, ΔΨK, возникают на границе двух металлов.
а) В случае, изображенном на схеме (рис. 14.7), это границы между проволокой из металла X, соединяющей электроды, и цинковой (ΔΨK,1) или медной (ΔΨK,,2) пластинкой.
б) Поскольку металлы характеризуются разной работой выхода электрона, то при прямом контакте металлов на их границе происходит некоторое перераспределение электронов.
в) Вновь обратим внимание на сумму двух скачков потенциала:
Здесь ΨX,1 и ΨX,2 — потенциалы проволоки в месте соединения с первым и вторым электродами.
г) Если бы во внешней цепи не было сопротивления (представленного, например, вольтметром), а сопротивлением проволоки можно было бы пренебречь, то потенциалы проволоки везде были бы одинаковы — в т. ч. на её концах (выполнялось бы равенство ΨX,1 = ΨX,2), и получалось бы, что
Это означает, что сумма контактных потенциалов не зависит от природы
проволоки X, а определяется только природой электродов.
5. а) В итоге измеряемая вольтметром ЭДС складывается из следующих шести скачков потенциала во внутренней части цепи:
б) Если мы подставим приводившиеся выше выражения для всех этих величин, то при условии (14.39,б) получим просто ноль, т. к. все потенциалы сократятся:
Таким образом, если во внешней цепи нет нагрузки (сопротивления), то элемент не вырабатывает ЭДС.
в) А теперь пусть во внешней цепи содержится сопротивление Rex. В этом случае потенциалы соединительной проволоки в местах контакта с электродами уже не одинаковы, и, как нетрудно убедиться путём такой же подстановки величин в (14.40), измеряемая ЭДС равна разности этих потенциалов:
если Rex ≠ 0 , то ЭДС = ΨX,2 – ΨX,1 . (14.41,б)
г) Причём данная разность максимальна тогда, когда во внешней среде не происходит падения напряжения:
при Rex → ∞ ЭДС → max.
6. Но не означает ли проведённое рассмотрение, что, благодаря влиянию диффузионных и контактных разностей потенциалов, максимальное значение измеряемой ЭДС всё-таки отличается от того значения (ΔΨрц), которое рассчитывается по энергии Гиббса? – Ни в коей мере.
I.. В отсутствие химической реакции для диффузионных потенциалов получаем:
т. к. потенциалы растворов не отличались бы друг от друга. Их различие
создается лишь благодаря тому, что растворы по-разному обмениваются с
металлическими пластинками ионами или электронами.
II. По той же причине равнялась бы нулю и сумма контактных скачков
потенциала (14.39,б), Таким образом, сумма всех скачков потенциала во вну-
тренней цепи элемента имеет своим происхождением только энергию ОВР.
Краткое содержание главы 14
В главе рассмотрены электродные процессы.
1. В ходе одного из них – ЭЛЕКТРОЛИЗА – происходит преобразование электрической энергии в энергию эндергонической ОВР.
2. Другой феномен – ГЕНЕРАЦИЯ ЭДС в гальванических элементах. Здесь, напротив, энергия ОВР преобразуется в электрическую энергию.
Так, в ЭЛЕМЕНТЕ ДАНИЭЛЯ-ЯКОБИ используется энергия следующей реакции:
3. а) При условии, что ток во внешней цепи — БЕСКОНЕЧНО МАЛ, возникающая ЭДС, или ЭЛЕКТРИЧЕСКИЙ ПОТЕНЦИАЛ реакции (∆Ψрц), однозначно связаны с энергией Гиббса:
б) Отсюда следует зависимость ЭДС от активностей участников (УРАВНЕНИЕ
НЕРНСТА) и от температуры:
4. а) С другой стороны, ∆ψрц можно представить и через ЭЛЕКТРОДНЫЕ ПОТЕНЦИАЛЫ:
где Ψ1 — потенциал катода (–), т. е. ОВ-пары, служащей ВОССТАНОВИТЕЛЕМ, а Ψ2 — потенциал анода (+), т. е. пары, служащей ОКИСЛИТЕЛЕМ.
б) Электродные потенциалы принято измерять по водородной шкале (при условии, что у водородного электрода P(H2) = 1атм и рН = 0). Тогда их называют РЕДОКС–ПОТЕНЦИАЛАМИ.
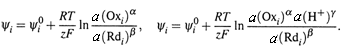 |
5. Редокс-потенциалы зависят от активностей участников соответствующей ПОЛУРЕАКЦИИ, например:
Эти выражения иногда называют уравнениями Нернста; в других источниках —
уравнениями Петерса.
6. а) СТАНДАРТНЫЕ значения ЭДС (∆Ψ°рц) и редокс-потенциалов (∆Ψ°i) относятся к ЕДИНИЧНЫМ АКТИВНОСТЯМ всех участников. Если среди них – и протоны, то данное условие означает, что рН = 0.
б) Используются также КАЖУЩИЕСЯ значения ЭДС (∆Ψ′рц), относящиеся к условию рН = 7,0.
7. а) В основе механизма генерации ЭДС – обмен ионами или электронами между электродом и раствором.
б) А измеряемая ЭДС складывается, по меньшей мере, из шести СКАЧКОВ ПОТЕНЦИАЛА во внутренней цепи элемента:
ЭДС = – (∆Ψ э, 1 + ∆Ψ э, 2 + ∆Ψ к, 1 + ∆Ψ к, 2 + ∆Ψ Д, 1+ ∆Ψ Д, 2 )
Эти скачки представляют собой разности потенциалов:
ЭЛЕКТРОДНЫЕ (∆Ψэ, i) — на границе металлических электродов с жидкой фазой полуэлементов;
КОНТАКТНЫЕ (∆Ψ к, i) — на границе электродов с соединяющей проволокой, и
ДИФФУЗИОННЫЕ (∆Ψ д, i) – на границе жидких фаз полуэлементов с содержимым cолевого мостика.
Глава 15. ВИДЫ ГАЛЬВАНИЧЕСКИХ ЭЛЕМЕНТОВ И
ЭЛЕКТРОДОВ. ПОТЕНЦИОМЕТРИЯ
Рассмотрев теоретические аспекты феномена генерации ЭДС в гальванических элементах, перейдем к более практическим вопросам.
В этой главе будут описаны различные виды этих элементов (до сих пор мы имели дело, фактически, только с элементом Даниэля–Якоби), а также соответствующие методы физико-химического анализа.
15.1. Обозначение электродов и элементов
1. а) Поскольку для аналитических и технических целей разработано много
разных электродов и элементов, используется специальная система их краткой записи. Вот, в частности, как записывают известные нам устройства:
I. водородный электрод –
II. элемент Даниэля–Якоби –
б) В общем виде запись произвольного гальванического элемента такова:
в) Нередко в этих обозначениях указывается природа металлических электродов (что уже видно в (15.2,а)) и солевого мостика, например:
2. Подобные записи основаны на следующей совокупности правил.
а) Первым пишут полуэлемент 1 (в соответствии с порядком нумерации, изложенным в п. 14.7), т. е. тот полуэлемент, на котором происходит полуреакция окисления и который поэтому служит донором электронов и является катодом.
б) Окисленные формы двух окислительно-восстановительных пар пишут в середине, а восстановленные формы — по краям.
в) Разные фазы одного полуэлемента разделяют вертикальной чертой.
г) Полуэлементы отделяют друг от друга или от солевого мостика двойной чертой. (Если между полуэлементами не указан солевой мостик, то их можно разделить одной пунктирной чертой.)
15.2. Классификация элементов по источнику энергии
1. До сих пор речь шла только о химических гальванических элементах, в
которых источником электрической энергии является энергия химической реакции (ОВР). Однако могут использоваться и концентрационные элементы.
а) Последние состоят из двух одинаковых по химической природе полу элементов, которые различаются концентрацией электролитов. Поскольку, согласно уравнению Нернста (14.22—14.24), электродный потенциал зависит от концентрации электролита, то полуэлементы в подобных элементах имеют различные потенциалы, а между ними устанавливается разность потенциалов ∆Ψ.
б) Таким образом, здесь источником ЭДС является энергия разности концентраций, т. е. концентрационная, или осмотическая энергия.
Примеры — элементы из двух медных или двух водородных электродов:
 |
I. Очевидно, выход ионов Сu2+ из медной пластинки легче происходит тогда, когда в растворе — меньшая концентрация этих ионов, и в этом случае на пластинке — больший отрицательный заряд. Отсюда следует, что катодом является полуэлектрод с меньшей концентрацией катионов в растворе и что электроны перемещаются отсюда на полуэлектрод с большей концентрацией. Таким образом, c1 < c2.
II. В случае водородного концентрационного элемента речь идет о концентрации (активности) протонов, т. е. ЭДС определяется разностью рН в полуэлементах. Действительно, проводя для произвольного водородного электрода
выкладки, подобные (14.34), получим (в В):
Поэтому ЭДС элемента (15.3,б) равна
2. а) Что же касается элементов химического типа, то для них иногда используют коэффициент полезного действия, определяя его следующим образом:
Здесь ∆Hрц— теплота реакции, а ∆Gрц — совершаемая за счет реакции работа (при бесконечно малом токе в элементе).
б) Однако известно (пп. 3.6 и 4.11), что бывают самопроизвольные процессы, в которых вклад в отрицательное значение ∆G дает не только энтальпийное, но и энтропийное слагаемое.
В таких случаях оказывается, что к. п.д., определенный по формуле (15.5),
больше 1. С моей точки зрения, такое определение неудачно.
15.3. Электроды первого рода
1. а) Классифицируют не только гальванические элементы, но и составляющие их электроды. Это тем более полезно, что в принципе электроды (полуэлементы), созданные на основе тех или иных ОВ–пар, можно объединять в
гальванический элемент в произвольной комбинации.
б) По природе компонентов и по принципу работы различают четыре группы электродов:
- электроды первого рода – M|M z+,
- электроды второго рода – КатАн↓М/Ан,
- редокс-электроды – (Pt) Ox|Rd,
- ионоселективные электроды.
2. а) В случае электродов первого рода компонентами являются простое вещество и простой ион этого вещества, что показывается обозначением M|M z+.
б) Некоторые примеры нам известны: электроды элемента Даниэля–Якоби
водородный электрод:
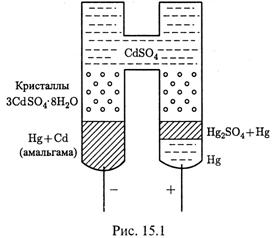 3. Еще важный пример: амальгамный электрод в элементе Вестона (левый электрод на рис. 15.1).
3. Еще важный пример: амальгамный электрод в элементе Вестона (левый электрод на рис. 15.1).
а) Амальгама — это раствор металла в ртути. В данном электроде используется раствор Сd в Hg. Над ним находятся кристаллы сульфата кадмия и насыщенный раствор этой соли.
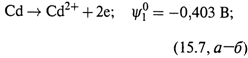 |
б) В электроде происходит полуреакция
как видно, она отвечает определению электродов 1 рода.
в) Однако имеется и особенность: поскольку кадмий находится не в твердом, а в растворенном состоянии, в уравнении Нернста, в отличие от (14.26), должна присутствовать и его активность:
4. а) Во втором полуэлементе элемента Вестона — паста из ртути и Hg2SO4, тоже покрытая кристаллами и насыщенным раствором сульфата кадмия. Здесь полуреакция такова:
б) Этот электрод называется ртутно–сульфатным и относится к электродам второго рода.
5. а) Элемент Вестона интересен тем, что обладает очень высокой стабильностью: его ЭДС не меняется в течение ряда лет, оставаясь равной
б) Поэтому его используют как элемент сравнения для измерения ЭДС других элементов.
6. а) Заметим, что у подавляющего большинства электродов первого рода потенциал, согласно уравнению Нернста, зависит от концентрации катиона, например:
б) Но бывают и электроды первого рода, чей потенциал зависит от концентрации аниона:
15.4. Электроды второго рода
1. а) Компонентами электродов второго рода являются
I. простое вещество (М),
II. его труднорастворимая соль (КатАн↓), а также
III. растворимый ион данной соли (Ан),
что и дает обобщенное обозначение, приведённое в п.15.3: КатАн↓M|Ан.
б) Подобный пример встречался выше в случае ртутно-сульфатного электрода в элементе Вестона (см. рис.15.1):
2. Другой пример — каломельный электрод (рис. 15.2):
Hg2Cl2|Hg(Pt)|Cl–.
а) Он содержит пасту, включающую каломель (Hg2Cl2), ртуть и KCl. Паста находится на чистой ртути и залита раствором KCl. Платиновый электрод погружен в ртуть. В этом полуэлементе происходит полуреакция, все компоненты которой находятся в пасте:
б) Если раствор KCl – насыщенный, потенциал электрода (в вольтах) таков:
 |
в) Здесь учтено, что активности твёрдой соли и жидкой ртути равны единице. Таким образом, потенциал зависит от концентрации ионов хлора, а по существу, от концентрации KCl в растворе над пастой.
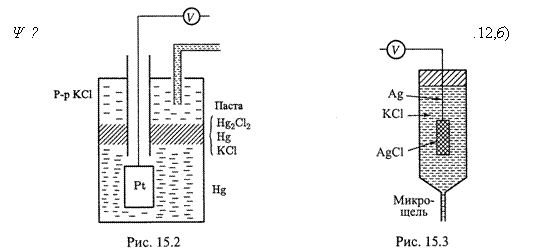
3. Еще один важный пример — хлорсеребряный электрод (рис. 15.3):
AgCl|Ag|Cl–.
а) Здесь серебряная проволока покрыта слоем AgCl и погружена в насыщенный раствор KCl. В ту или иную сторону происходит полуреакция:
б) В случае прямого её направления образующийся металл оседает на проволоке, а ион Cl– переходит в раствор, вследствие чего на металлическом электроде создается дефицит электронов.
в) Потенциал вновь зависит от концентрации ионов хлора:
4. а) Итак, потенциалы электродов второго рода зависят от концентраций
анионов.
б) А стандартное значение такого потенциала связано со стандартным потенциалом пары M+/М, где М — простое вещество, входящее в состав рассматриваемого электрода.
5. Покажем эту связь на примере хлорсеребряного электрода.
а) При его функционировании (как и в случае любого электрода второго рода) сочетаются два процесса:
I. растворение плохорастворимой соли AgCl с одновременной диссоциацией на ионы, характеризуемое произведением растворимости:
Пр = а(Ag+)∙а(Cl–) = 1.78∙10–10 , (15.15,а)
II. и восстановление иона:
Ag+ + e → Ag , где Ψ0(Ag+/Ag) = 0,799 В. (15.15,б)
Следовательно, потенциал хлорсеребряного электрода – это, по существу, потенциал пары Ag+/Ag:
Ψ(AgCl/Ag/Cl–) = Ψ(Ag+/Ag) . (15.16)
б) Однако стандартные потенциалы данных систем различны: Ψ0(Ag+/Ag) соответствует условию а(Ag+) = 1, а Ψ0(AgCl/Ag/Cl–) – условию а(Cl–) = 1. Действительно, уравнения Нернста имеют вид:
I. Ψ(Ag+/Ag) = Ψ0(Ag+/Ag) + 0,059∙lg a(Ag+) =
= Ψ0(Ag+/Ag) + 0,059 ∙ lg [Пр /а(Cl–)] , (15.17,a)
II. Ψ(AgCl/Ag/Cl–) = Ψ0(AgCl/Ag/Cl–) – 0,059∙lg a(Cl–) . (15.17,б)
в) Приравнивая, в соответствии с (15.16), эти выражения, находим стандартный потенциал хлорсеребряного электрода:
Ψ0(AgCl/Ag/Cl–) = Ψ0(Ag+/Ag) + 0,059 ∙ lg Пр ≈
≈ 0,799 В – 0,576 В ≈ 0,223 В. (15.18)
г) Таким образом, здесь исходный стандартный потенциал (потенциал пары Ag+/Ag) сильно уменьшается за счёт того, что соль AgCl, участвующая в образовании данной пары, очень плохо растворима.
6. Хлорсеребряный и каломельный электроды часто используются (вместо водородного электрода) в качестве электрода сравнения (не путать с элементом сравнения, например, элементом Вестона).
Для этого какой-нибудь из них включают в цепь с электродом, чей потенциал надо измерить. Определив в таком гальваническом элементе ΔΨ и зная стандартный потенциал электрода сравнения, нетрудно найти и искомый потенциал. Так, если электрод сравнения выступает в качестве окислителя, то
ΔΨ = Ψсрав – Ψх, откуда Ψх = Ψсрав – ΔΨ . (15.19,а-б)
15.5. Окислительно-восстановительные (или редокс-) электроды
1. а) В принципе, каждый электрод является окислительно-восстановительным. Но под термином «редокс-электроды» понимают такие полуэлементы, где все компоненты полуреакции (Ох и Rd) находятся в растворе; металлические же электроды, погруженные в раствор, в реакции не участвуют, а служат лишь переносчиками электронов. Именно о таких электродах шла речь в п. 14.4, где была приведена и общая схема элемента из двух редокс-электродов (см. рис. 14.3).
б) Компонентами ОВ-пары при этом могут быть неорганические вещества,
например:
Однако для органических веществ редокс-электроды особенно важны, т. к. яв-
ляются единственным способом образовать полуэлемент.
2. а) Наиболее известный пример — хингидронный электрод. Здесь
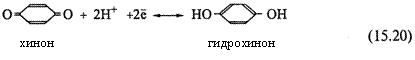 |
платиновая пластинка погружена в насыщенный раствор хингидрона в присутствии твердого хингидрона. Растворенный хингидрон диссоциирует на эквивалентные количества хинона (Ох) и гидрохинона (Rd), связанные реакцией восстановления:
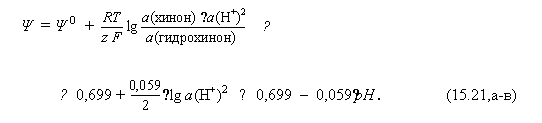 |
б) В данной полуреакции фигурируют протоны; поэтому потенциал электрода зависит от рН:
Здесь учтено, что z =2 и что концентрации хинона и гидрохинона в растворе практически одинаковы в силу эквимолекулярного образования их из хингидрона.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 |
