
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Диффузия газов и альвеолярно-капиллярная блокада. Переход газов из альвеол в крови и обратно представляет собой диффузию газов через проницаемую для них мембрану: молекулы газа переходят из зоны высокого в зону низкого парциального давления. Следовательно, у человека диффузия может быть выражена объемом газов (например, О2)г который способен пройти через альвеолярно-капиллярную мембрану за 1 мин при градиенте парциальных давлений, равном 1 мм рт. ст. У здорового человека диффузионная способность легких для О2 составляет 15—20 мл/(мин · мм рт. ст.). Эта величина возрастает при физической нагрузке. Через всю поверхность здоровых легких в организм может проникнуть более 6 л О2 в минуту. Обычно в клинической практике эту величину не определяют, поскольку результаты, полученные при ее расчете, сугубо ориентировочны.
J. Austrian и соавт. (1951) назвали нарушение диффузионной способности легких альвеолярно-капиллярной блокадой. Они обратили внимание на то, что синдром альвеолярно-капиллярной блокады неспецифичен. Он возникает при тяжелых заболеваниях и поражениях легких. Патогенез этого явления сложен и до конца не изучен. Во всяком случае, стало очевидным, что синдром альвеолярно-капиллярной блокады может быть обусловлен многочисленными причинами. В пользу этого говорит сложность структуры альвеолярно-капиллярной мембраны, обусловливающая многоступенчатость самого процесса переноса газов. Помимо основного фактора простой физической диффузии, в процессе переноса газов через мембрану участвуют и факторы, активирующие его и связанные с наличием в альвеолярно-капиллярной мембране особых образований — везикул, ускоряющих активный перенос веществ через мембрану, а также с наличием белков и липидов особой структуры, выстилающих поверхность алвеолы.
Компоненты патологии легочной ткани при СДРВ, которые определяют нарушение диффузии газов через альвеолярно-капиллярную мембрану, хорошо известны, хотя роль каждого из них мало изучена. Наиболее значимыми из этих компонентов являются интерстициальный (перикапиллярный) отек, гипертрофия альвеолярных клеток, образование гиалиновых мембран внутри альвеол и интерстициальный фиброз легких. С клинических позиций все четыре компонента следует рассматривать вместе как общую причину возникающего увеличения дистанции прохождения молекул О2 и СО2 и увеличения барьера между пространством альвеолы и эритроцитарным гемоглобином. В конечном счете парциальное давление газов в крови зависит от «дистанции пробега» и от разности парциальных давлений газа по обе стороны мембраны. С физиологической точки зрения в нормальных легких диффузия О2 через альвеолярно-ка-пиллярную мембрану имеет широкие пределы. Если у больного с увеличенной «дистанцией пробега», вызванной образованием тканевого легочного барьера, применять 100% О2, т. е. увеличить разность парциальных давлений O2 между альвеолярным газом и кровью, то можно снизить «диффузионные потери». Однако больной должен долго дышать 100% О2, чтобы достичь полного вымывания N2 из образовавшихся альвеолярных ловушек. Практически в начальных фазах СДРВ для этого требуется не менее 30—45 мин.
Синдром капиллярного просачивания и отек легких. Развитие интерстициального и альвеолярного отека с повышением левопредсердного или легочного венозного давления является следствием закона Старлинга, определяющего условия транскапиллярного жидкостного обмена. Механизмы, ответственные за развитие отека легких при нормальном легочном венозном давлении (так называемого отека с низким легочным давлением), достаточно сложны. Обычно называют три причины, обусловливающие отек: снижение онкотического давления плазмы, повышение легочной капиллярной проницаемости, изменение функции легочных лимфатических сосудов [Sta-ub N. С., 1974]. Первые два механизма часто комбинируются и приводят к увеличению скорости лимфообращения и повышению концентрации белков в лимфе легких [Demling R. H. et al., 1979]. Роль лимфы в поддержании нормальной анатомии и функции интерстициального легочного пространства исключительно велика. Однако массивные инфузии жидкостей, развитие инфекции, введение лекарств и изменения внутрилегочного давления существенно влияют на лимфатическую систему легких и могут способствовать возникновению отека их~. Если возникает функциональная блокада тока лимфы в сочетании с выраженным повышением внутрибронхиального давления, то в неподатливых легких жидкость быстро накапливается в легочном ин-терстиции даже при относительно малом изменении онкотического давления или легочной мембранной проницаемости.
ОДН, которая развивается в результате синдрома капиллярного просачивания и отёка легких, может быть двух типов. ОДН первого типа характеризуется развитием так называемого влажного легкого и оценивается сейчас как более благоприятное состояние. На фоне интерстициального отека легких определяется нормальное легочное капиллярное давление (судят по результатам исследования давления заклинивания легочной артерии) и отсутствует легочная гипертензия. Диуретическая терапия с использованием фуросемида, ультрагемофильтрация или ограничение жидкостной нагрузки достаточно эффективны.
При ОДН второго типа течение болезни более тяжелое, прогноз неблагоприятен. Главным патологическим симптомом является легочная гипертензия на фоне интерстициального отека легких. Легочная ангиография не выявляет легочных капилляров, которые при этом варианте бывают заполнены фибрино-выми (иногда эритроцитарными) микроэмболами. Обычно подобное состояние сочетается с выраженным ДВС-синдромом, преимущественно с его первой фазой (гиперкоагуляции). ОДН развивается стремительно и характеризуется тяжелой гипоксе-мией, обусловливающей необратимость заболевания. При этом, помимо мероприятий по поддержанию адекватной оксигенации крови (ИВЛ или как крайней меры — экстракорпоральной мембранной оксигенации), показано лечение с использованием стрептокиназы и гепарина.
Патология перфузии легких. Нарушения вентиляции при СДРВ происходят одновременно с расстройствами кровообращения в легких. Наиболее отчетливые нарушения легочного кровообращения развиваются главным образом в венозной системе и выражаются преимущественно в тромбоэмболии; при этом кровь механически шунтируется в неповрежденные сосудистые зоны. Тромбоциты в сгустках крови начинают высвобождать факторы, которые вызывают бронхоконстрикцию во всех зонах легких, что ведет к углублению синдрома «промахивания» и разобщению вентиляции и перфузии. Те же процессы, хотя и менее выраженные, наблюдаются в эмболах, возникающих в результате многократных переливаний крови. Наиболее общей причиной перераспределения легочного кровотока является левожелудочковая недостаточность с повышением давления в левом предсердии. При этом повышенное легочное венозное давление способствует увеличению легочного кровотока в области плохо вентилируемых зон легких и таким образом увеличивает шунтирование. Увеличение количества внесосуди-стой жидкости в легких, вызванное повышением легочного капиллярного давления, содействует закрытию малых дыхательных путей и коллапсу альвеол.
Другие причины увеличения легочного шунтирования при СДРВ включают механизмы, важность которых несомненна, но которые с клинических позиций бывает трудно оценить. Первый из них связан с ускорением пассажа эритроцита через легочные капилляры. Как известно, у большого числа больных на первых этапах развития СДРВ наблюдается гипердинамический синдром, который характеризуется очень высоким сердечным выбросом. Обычно регистрируют увеличение сердечного выброса в 2—3 раза в ранних стадиях после начала лечения. Особенно выражен этот синдром у больных, состояние которых осложнено сепсисом. С позиций физиологии сердечный выброс должен быть приспособлен по объему к уменьшенному числу легочных капилляров. В результате продолжительность среднего транзитного времени для каждого, эритроцита существенно снижается. Очевидно, что при таких обстоятельствах, особенно если усилен диффузионный барьер или имеется гиповентиляция, развивается неполная оксигенация гемоглобина, что может восприниматься как увеличение легочного шунтирования [West J. В., 1974]. Этот эффект еще более усиливается, если кровь, проникающая в легкие, имеет ацидотическую реакцию, которая смещает кривую оксигенации гемоглобина вправо.
Гипервентиляция и гипокапния. В реаниматологической практике высокий минутный объем дыхания (MOB) называют «гипервентиляцией». Этим термином можно охарактеризовать не только спонтанное дыхание больного, но также режим ИВЛ. С точки зрения физиологии гипервентиляция (одышка) представляет собой усиленный режим дыхания, обусловленный активацией дыхательного центра под влиянием изменившихся условий внутренней среды в организме, в частности при снижении РаО2 при возникновении ацидоза или повышении температуры тела.
Нарушения ритма и интенсивности дыхания являются также нормальной реакцией на тяжелую травму, боль, страх, раздражение брюшины. С учетом этого, по-видимому, более правильно не столько искать внутреннюю причинную связь между возникающей гипервентиляцией и биохимическими изменениями внутренней среды (хотя это тоже необходимо), сколько принять к сведению факт гипервентиляции как первого симптома развивающейся дыхательной недостаточности.
Ранний период развития дыхательных расстройств у больных в критическом состоянии характеризуется гипервентиляцией, приводящей к гипокапнии и умеренному респираторному алкалозу. Подобная спонтанная гипервентиляция может, по-видимому, иметь отношение к этиологическому фактору, т. е. может быть вызвана травмой, кровопотерей или инфекционным фактором лишь в самом начале.
Не существует убедительного физиологического объяснения раннего появления гипервентиляции. Однако известно, что в одних случаях это происходит на фоне ранней неадекватной оксигенации или позже, при полном развитии СДРВ, когда гипервентиляция может быть следствием развивающейся гипоксии, и тогда ее появление кажется вполне закономерным. В других случаях (пожалуй, в большинстве) гипервентиляция появляется без признаков гипоксии. Выраженная, гипокапния и обусловленный ею алкалоз нежелательны всегда, особенно у больных с пороками сердца, периферической сосудистой недостаточностью, аритмией сердца или при лечении дигиталисом. Поскольку гипокапния и дыхательный алкалоз — непременное следствие и аппаратной гипервентиляции, целесообразно по возможности предупреждать развитие этого состояния увеличением объема мертвого пространства с помощью дополнительной вставки между эндотрахеальной трубкой и респиратором. Алкалоз плохо переносится человеком. Его кажущаяся безвредность в противоположность ацидозу частично является результатом логарифмической природы рН-шкалы: равные изменения рН при алкалозе сопровождаются меньшими изменениями [Н+], чем при ацидозе.
Респираторный алкалоз оказывает существенное влияние на деятельность мозга. Этот вопрос был изучен J. Hinshaw и R. Booth (1941) при оценке деятельности пилотов в реальных условиях. При исследовании реакции пилотов в стрессовых ситуациях авторы обнаружили, что гипервентиляция нарушает точность различных производимых пилотами привычных процедур и объясняли это церебральной ишемией. S. S. Kety и С. F. Schmidt (1946) в хорошо известных теперь исследованиях, определяя изменения церебрального кровотока под влиянием изменений Рсо2, установили, что при спонтанной гипервентиляции у добровольцев, приводившей к снижению Рсо2 до 18 мм рт. ст., мозговой кровоток уменьшался на 32%. Продолжением этих исследований явились работы J. S. Meyer и F. Gotoh (1966), в которых в экспериментах на кошках было показано, что гипервентиляция, снижавшая Рсо2 до 10 мм рт. ст., существенно снижала Ро2 в коре мозга. При этом возникали также гистологические изменения в мозге.
Другие эффекты, вызванные респираторным алкалозом, выражаются в вазоконстрикции, снижении артериального давления, повышении числа циркулирующих эритроцитов при снижении общего объема циркулирующей плазмы, наконец, снижением концентрации Са2+ при повышении содержания общего кальция плазмы [Robinson J. S., Gray T. С., 1961].
Расстройства периферического кровообращения и лактат-ацидоз. Уже на ранних стадиях развития синдрома дыхательных расстройств наблюдается умеренное повышение концентрации L—. Обычно развивающийся лактат-ацидоз сочетается с низким сердечным выбросом, начальным дыхательным алкалозом и гипокапнией. Во второй фазе синдрома можно наблюдать нормализацию уровня L— в крови. Однако с переходом в третью и особенно в четвертую фазу вновь можно отметить повышение содержания L— в крови и лактат-ацидоз, которые развиваются теперь на фоне расстройств системного кровообращения. Подобная динамика содержания l - в крови и ацидоза характерна для больных с гиподинами-ческим синдромом. Однако описанную динамику L— крови вряд ли можно объяснить лишь системной циркуляторной недостаточностью и самим СДРВ, обусловливающими гипоксию. По-видимому, здесь действуют более сложные и неоднозначные факторы. Повышение уровня L— в крови наблюдается, в частности, при гипервентиляционной гипокапнии и в отсутствие существенных нарушений системного кровообращения. При этом содержание L— в крови быстро нормализуется при прекращении гипервентиляции [Robinson J. S., Gray Т. С., 1961]. Описано также повышение концентрации l - в крови после введения NaHCO3 [Haldi H., 1933].
W. Huckabee (1958), а затем его последователи показали, что пропорционально повышению L~ в крови повышается и уровень пирувата.
Лактат является нормальным метаболитом глюкозы, появляющимся в результате тканевого анаэробного гликолиза. При образовании L—появляется эквимолярное количество Н+. Это и определяет возможный сдвиг КОС в сторону ацидоза. Образование L— не дает организму существенной энергетической прибыли: большая часть калорического выхода при метаболизме глюкозы получается путем окислительного фосфорилирования.
Повышение уровня L— в первой фазе СДРВ бывает обычно умеренным (до 2—3 ммоль/л) и наблюдается 2—3 дня. Если кровообращение при этом стабилизируется, то большая часть L— может метаболизироваться и уровень его постепенно приходит к норме. При прогрессировании СДРВ с увеличением венозного примешивания и углублением артериальной гипоксе-мии как при гиподинамии, так и при гипердинамии гипоперфузия тканей усиливается. Концентрация L— продолжает повышаться и в ряде случаев достигает 4—6 ммоль/л. Обычно это сопровождается высоким отношением лактат/пируват.
Таким образом, системная гипоперфузия — наиболее важная причина лактацидемии. Другая очевидная причина лактат-ацидоза — артериальная гипоксемия — при тех величинах РаO2, которые допускают жизнеспособность тканей, как свидетельствуют данные W. Huckabee (1958), в общем не ведет к избытку продукции лактата. Все же лактацидемия является достаточно надежным индикатором гипоксии организма. При этом гипоксию следует рассматривать как глобальное явление для организма, возникающее в результате не одного, а множества факторов.
Возможность охарактеризовать гипоксию по концентрации лактата крови была использована W. Huckabee (1958). Автор взял за основу уравнение восстановления пирувата в лактат при участии систем NAD/NADH и вывел формулу, позволившую вычислить тот «избыточный» лактат, появление которого не объясняется исходной концентрацией лактата и пирувата. Этот показатель обозначает как ExL. Концепция W. Huckabee широко распространилась и используется в научных изысканиях по проблеме гипоксии.'По мнению этого автора и других исследователей, показателей ExL наилучшим образом коррелирует с другими показателями, характеризующими гипоксию, и с клиническими данными.
Однако концепция W. Huckabee вызвала также и критику. Основные аргументы возражающих сводятся к указаниям на отсутствие равновесных состояний реакций в живом организме, на различное содержание лактата и пирувата внутри и вне клетки, а также на изменения константы диссоциации. При сниженной перфузии тканей в кровеносное русло попадает большое количество L—, особенно из скелетных мышц. В норме печень и миокард способны к самоочищению от L—, т. е. могут активно метаболизировать его. При расстройствах периферического кровообращения, малом сердечном выбросе и в условиях гипоксемии это становится невозможным. При концентрации L,- в крови свыше 10 ммоль/л больные не выздоравливают. При этом почти весь лактат (свыше 80% его) определяется в виде ExL. В отсутствие почечной недостаточности или диабетического ацидоза некоторое снижение BE может объясняться, в частности, и повышением концентрации l - в крови. Таким образом, уровень лактата в крови можно считать прогностическим критерием.
Другим (негипоксическим) источником образования L - в организме является молочно-кислое брожение, которое происходит с участием микроорганизмов в анаэробных условиях; это нередко наблюдается у обожженных. В клинических условиях иногда используют также растворы, содержащие L—. В ряде случаев растворы лактата предпочтительнее растворов, например, гидрокарбоната натрия. Но если раствор, содержащий l-, дается больному, который не в состоянии метаболизировать собственный избыточно образующийся L—, то возникает лактат-ацидоз. Переливание фруктозы, а также введение больших доз норадреналина и изупрела повышает содержание l - в крови.
Должная клиническая оценка лактат-ацидоза может быть сделана при сопоставлении его с другими видами ацидоза. Клиницистам известно, что остро развившийся дыхательный ацидоз, достигающий по величине рН уровней, близких к терминальным, хорошо переносится организмом короткое время.
Хронический метаболический ацидоз с рН, близким к 7,25—7,20, часто наблюдаемый у больных с хронической почечной недостаточностью или диабетом (у больных, состояние которых очень тяжелое), не является смертельным и может длительно переноситься организмом.
Совершенно иначе обстоит дело с ацидозом, развивающимся в условиях низкого сердечного выброса, особенно если гипоперфузия сочетается с артериальной гипоксемией, т. е. у тех больных, которых мы называем критическими. Это связано с тем, что при низком сердечном выбросе, в частности при шоке любого происхождения, лактат-ацидоз возникает в самой клетке и, таким образом, касается всего организма в целом. Если почечный ацидоз обусловлен плохим выведением Н+ при нормальной скорости их образования в организме, то ацидоз при тканевой гипоперфузии характеризуется повышенной продукцией Н+ в условиях анаэробиоза и отражает нарушение метаболизма в клетке, в частности в митохондриях. При этом типе ацидоза создаются благоприятные условия для развития брадикардии и асистолии.
Метаболический алкалоз. Вместе с гипоксемией метаболический алкалоз является важнейшей причиной, формирующей критическое состояние больного при СДРВ. Если такое состояние вызвано продолжительным угнетением системного и органного кровообращения, т. е. если в основе критического состояния лежит длительный «синдром малого выброса», то чаще всего развивается выраженный компенсированный или декомпенсированный (в зависимости от обстоятельств и выраженности синдрома) ацидоз. В большинстве случаев в клинических условиях удается корригировать гипоперфузионный синдром, используя обычный комплекс мер реанимации и интенсивной терапии. Фаза метаболического ацидоза в этих случаях оказывается непродолжительной и вскоре переходит в метаболический алкалоз. Этому обычно предшествуют проявления начинающейся ОДН.
К дыхательному алкалозу, происхождение которого в ранних стадиях ОДН связано исключительно с гипервентиляцией, присоединяется алкалоз метаболического характера. F. D. Мооге и соавт. (1969) считают, что в тех случаях, когда состояние низкого кровотока достигает крайних степеней и ведет к смертельному исходу с поздним лактат-ацидозом, самым первым проявлением нарушений КОС организма служит комбинация дыхательного и метаболического алкалоза, который вначале выражается лишь в незначительном избытке оснований в крови. Одной из причин его возникновения является метаболизм цитрата натрия, попавшего в кровь больного при массивных гемотрансфузиях, обычных для подобных больных.
Однако заметный избыток НСО3— в организме может накапливаться лишь тогда, когда степень его экскреции почками значительно снижается. Именно такая ситуация возникает у больных в критическом состоянии, когда клубочковая фильтрация у них угнетается. Сопутствующим фактором является развивающийся при критическом состоянии вторичный гиперальдо-стеронизм. Непосредственной причиной его является стрессовое состояние и почечная гипоперфузия, вызывающая активацию ренин-ангиотензин-альдостероновой системы организма. Гиперальдостероновая фаза сочетается с исключительно высокой канальцевой реабсорбцией Na+ и упорной парадоксальной ацидурией, несмотря на плазменный алкалоз. Ацидурия (на первый взгляд, не сочетающаяся с алкалозом) является, как правило, прогностически неблагоприятным признаком. Нарборот, переход кислой реакции мочи в щелочную при упорном метаболическом алкалозе у больного в критическом состоянии оценивается как признак появления натрийурической фазы, обещающей скорое выведение избытка Na+ и, следовательно, ликвидацию алкалоза.
Важен еще один клинический аспект алкалоза—его влияние на кислородный баланс организма. Обычно больной с выраженным метаболическим алкалозом имеет хороший внешний вид: кожа теплая, розового цвета, без признаков цианоза, периферические сосуды достаточно расширены. Однако часто это сочетается с неспокойным поведением, иногда возбуждением и недостаточным контактом с медицинским персоналом. Нередко это бывает единственным клиническим признаком тяжелого состояния. Отрицательное влияние метаболического алкалоза в подобных случаях выражается прежде всего в его воздействии на диссоциацию оксигемоглобина: под влиянием высокого рН крови сродство гемоглобина к О2 значительно увеличивается и, следовательно, ухудшается отдача О2 гемоглобином в периферических тканях (эффект Бора). Единственными факторами, положительно воздействующими в таком случае на способность гемоглобина отщеплять кислород (за исключением 2,3-ДФГ, повышения температуры тела), являются накопление кислот в, организме и снижение рН. Таким образом, при алкалозе, несмотря на достаточно высокое насыщение гемоглобина О2, периферические ткани организма страдают от недостатка кислорода.
Терминальная гиперкапния. В развитии заболевания наступает период, когда нарушение отношения вентиляция/перфузия достигает наивысшего уровня и к тяжелой гипоксемии присоединяется гиперкапния. Механизм ее развития совпадает с механизмом гипоксемии: контакт вентилирующего газа с кровью и легких из-за разобщения газового и кровяного потоков невозможен. Отмечаются низкие величины Рсо2 выдыхаемого газа (реCO2.) и очень высокие цифры РаCO2. К факторам, способствующим гиперкапнии, относятся также обширные воспалительные процессы. В описанных ситуациях смерть наступает быстро.
Формирование легочного шунта. СДРВ у больных в критическом состоянии характеризуется прогрессирующей артериальной гипоксемией, которая наблюдается и в ранние, и в поздние сроки заболевания. Как уже указывалось, в начальных стадиях она сочетается с гипервентиляционной гипокапнией. Подобную комбинацию можно наблюдать несколько дней. Однако при неблагоприятном развитии заболевания гипоксия начинает сочетаться с гиперкапнией, и это является опасным прогностическим признаком. Другой характерной чертой гипоксемии является ее устойчивость к ингаляции 100% О2. В подобной драматической ситуации альвеолярно-артериальная разность Ро2 [P(A-a)O2] достигает 400—500 мм рт. ст. Это означает, что поражение легких имеет не только функциональный, но и анатомический характер: легкие не способны осуществлять адекватный газообмен. В возникновении высокой Р(А-а)Ог при ингаляции 100% О2 основную роль играет возникновение патологического и увеличение физиологического (т. е. прямых вено-артериальных сообщений нормальными каналами) легочного шунта. При меньших FiO2 могут иметь значение также нарушения равномерности распределения коэффициентов вентиляция/кровоток, а также, возможно, поступление в легкие крови с низким содержанием кислорода. Поскольку роль диффузии в клинической практике оценить очень трудно, а применение высоких концентраций кислорода делает нарушения диффузии маловероятной причиной гипоксемии, эти нарушения обычно не принимают во внимание как причину снижения РаO2 при СДРВ.
Таким образом, патогенетической сущностью ОДН при критических состояниях является веноартериальное примешивание, вызванное вентиляционно-перфузионным дисбалансом и легочным шунтированием. Конечным результатом нарушений легочного газообмена при СДРВ, обусловливающих состояние больного, является гипоксия.
Мы рассматривали подробно механизмы шунтирования крови в легких. Считаем необходимым напомнить, что у здоровых людей некоторое количество венозной крови с насыщением гемоглобина О2 около 70% поступает в левый желудочек сердца, не оксигенируясь. Это нормальный физиологический шунт, который образован прохождением крови через необширные, мало вентилируемые зоны легких, через бронхиальные сосуды и, возможно, некоторой частью коронарного кровотока. Общее количество оксигенированной в легких крови обычно менее 2 % сердечного выброса. Этот объем выявляется при ингаляции 100% О2 в течение 30 мин или более (проба Уленбрука). Предельно достигаемое РаОз при дыхании 100% О2 при давлении 1 атм (760 мм рт. ст.) в норме составляет 550—600 мм рт. ст. Теоретически с коррекцией на водные пары и СО2 оно должно быть около 680 мм рт. ст.
Что же является патофизиологическим механизмом, приводящим к прохождению 50—60% сердечного выброса через легкие без оксигенации в них, которое так часто отмечается у больных в крайне тяжелом, критическом состоянии?
С позиции патофизиологии это вентиляционно-перфузионный дисбаланс. Именно он является главной причиной повышения легочного шунта у наших пациентов. Возможны варианты подобного дисбаланса от сочетания хорошей перфузии с плохой вентиляцией или полным отсутствием вентиляции некоторых зон легких до сочетания высокой вентиляции с плохой перфузией или отсутствием ее.
Участки легких с плохой вентиляцией обязательно должны быть перфузируемы, чтобы проявился шунт., Если перфузия прекращается, то в этой зоне нет и шунта. Классические эксперименты провели в этом направлении Н. Nilsson и соавт. (1956), которые использовали трубку Карленса, чтобы путем окклюзии главного бронха выключить легкое из вентиляции. Вся проходящая через выключенную долю кровь попадала в левое предсердие неоксигенированной. В результате степень артериальной гипоксемии не снижалась даже при ингаляции 100% О2. Затем, когда кровоснабжение этой доли легкого прекращалось и невентилируемая доля переставала перфузироваться, артериальная оксигенация восстанавливалась до нормы.
Клинический эквивалент подобного эксперимента, ведущий к снижению вентиляции перфузируемых зон легких, по-видимому, может быть обусловлен тремя механизмами: констрикцией или частичной обструкцией больших бронхов, закрытием малых дыхательных путей и облитерацией альвеол. Сужение крупных дыхательных путей может быть вызвано раздражением их какими-либо ингалируемыми веществами, спазмирующими агентами, появляющимися при эволюции тромбоцитов, лекарственными средствами, нейроэндокринными реакциями организма. Подобная бронхоконстрикция или бронхообструкция может быть также обусловлена большим количеством недренируемого бронхиального секрета или какого-либо экссудата, который затем вызывает сужение просвета бронхов.
Концепция патологического венозно-артериального шунта была впервые предложена S. Berggren (1942). Согласно этой концепции:
QT = Qc + Qs,
где Qc — легочный капиллярный кровоток; Qs —кровоток через шунт; qt — сердечный выброс.
Общее уравнение шунта может быть выражено через содержание кислорода [Rahn H., Fahri L., 1964].
где Qs — общий легочный шунт, Qt — сердечный выброс, ССО2 – содержание О2 в легочных капиллярах, СаО2 —артериальной крови, СVO2, — смешанной венозной крови соответственно.
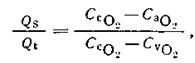 (1)
(1)
В клинических условиях удобно использовать уравнение, в котором СCO2 выражено через РА02:
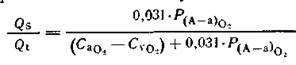 (2)
(2)
где 0,031 —коэффициент растворимости О2 в плазме, мД/(л-мм рт. ст.). Для вычисления раО2 используют формулу:
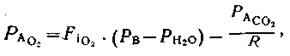 (3)
(3)
где Рв — атмосферное давление, Рн2о— парциальное давление (47 мм рт. ст.) водяных паров при 37°С, РАСО2—Рсо2 в альвеолярном газе, принимаемое равным PaСО2 P—дыхательный коэффициент, обычно принимаемый равным 0,8.
Уравнение (2) справедливо лишь при высоких РАО2 и РаО2, т. е. для тех случаев, когда насыщение гемоглобина О2 близко к 100%. У тяжелобольных с дыхательной недостаточностью, у которых это условие не выполняется, уравнение (2) может дать ошибочные результаты.
F. D. Moore и соавт. (1969) считают, что если РаО2 выше 100 мм рт. ст., то (независимо от FIO2) применять уравнение (2) можно. Первый расчет целесообразно сделать с использованием 100% О2 во вдыхаемой смеси. Вычисление легочного шунта по указанным показателям можно трактовать как тест на тяжесть поражения легких.
Наиболее точно легочный шунт можно определить при использовании катетера Свена — Ганса.
Эффективность вентиляции легких. В современной медицине для оценки легочного газообмена определяют Рао2 и Расо2. Согласно современным концепциям, Paо2, обратно пропорционально объему легочной вентиляции: чем меньше MOB, тем выше Рао2. Справедливость этой концепции применительно к здоровому организму была много раз доказана и не вызывает сомнений. В условиях дыхательной недостаточности закономерности значительно меняются. J. West (1971) установил, что Расо2 как и РаО2 находится в зависимости от вентиляционно-перфузионных отношений в легких и существенно меняется при поражении легких.
Прямое измерение и оценка Ра02 и насыщения гемоглобина O2 являются единственными способами оценки вентиляции легких. При постоянных концентрациях О2 во вдыхаемой смеси и pvq, изменения РаO2 зависят от нарушений отношения вентиляция/перфузия. Величина Ра02 дает возможность оценить в целом адекватность газообмена через альвеолярно-капиллярную мембрану. Другими словами, этот показатель дает возможность определить наличие или отсутствие в легких областей, где перфузия преобладает над вентиляцией. Если имеется информация о рао2 и о Рао2. то есть возможность рассчитать разность этих величин как показатель адекватности функции легких. При дыхании воздухом в условиях нормального атмосферного давления, Ро2 которого 159 мм рт. ст. (или около 150 мм рт. ст. с учетом увлажнения воздуха в легких), фактическое рао2 должно составлять 100 мм рт. ст.; при этом Ро2 равно 95—90 мм рт. ст. При дыхании смесью с FiО2, 0,5 (50% О2) Ро2 которой составляет около 350 мм рт. ст., Ра02 у здорового человека должно быть около 280—290 мм рт. ст. (за вычетом Рсо2 и Рн2о).
Патоморфология. При вскрытии погибших в результате выраженного СДРВ выявляется большое разнообразие патоморфологических данных, но имеется также множество сходных проявлений. Это связано с тем, что независимо от первичной этиологии в развитии СДРВ при критических состояниях имеется много общих патогенетических факторов. Из них наиболее важны интерстициальный отек с увеличением толщины межальвеолярных перегородок, прогрессирующий интраальвеолярный отек и наличие гиалиновых мембран в альвеолах.
Масса легких превышает нормальную в 3—4 раза, они кажутся раздутыми. На поверхности плевры можно видеть кровоизлияния и небольшие зоны ателектазов. Поверхность разреза легких обычно сочная, на ней также видны мелкие кровоизлияния; в остальных участках цвет легких остается нормальным. Часто наблюдаются участки пневмонии различных размеров, более яркие, чем окружающая паренхима. Почти всегда выражен трахеобронхит. Слизистая оболочка трахеи и бронхов темная. При наличии трахеостомы в ее зоне выражена воспалительная реакция.
Микроскопически можно выявить интерстициальный отек, который проявляется расширением альвеолярных стенок с выбуханием участков ткани в просвет альвеол. Видны также гиалиновые и фибринозные конгломераты на альвеолярных стенках и в альвеолах, фокальные альвеолярные и интерстициальные кровоизлияния. Определяются гиперплазия и гипертрофия альвеолярных выстилающих клеток. На истонченных альвеолярных перегородках можно видеть коллаген (рис. 4.2, а). В поздних стадиях коллагеновая выстилка эволюционирует до выраженного интерстициального фиброза, который сморщивает альвеолы и закупоривает их (рис. 4.2,6).
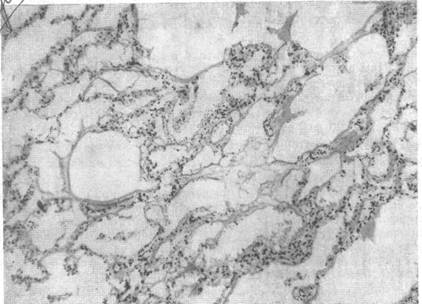
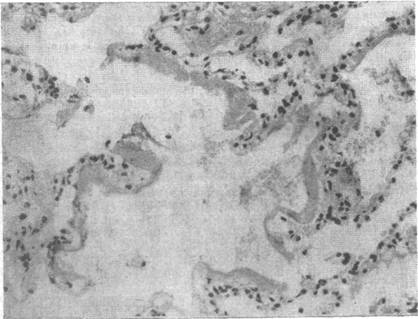
Рис. 4.2. Патоморфологическая картина легких при СДРВ. Х200. Окраска гематоксилин-эозином.
а — альвеолы расширены, в просвете их пристеночные гиалиновые мембраны, имеются альвеолярные геморрагии, межальвеолярные перегородки утолщены; б — массивные накопления гиалиновых мембран в альвеолах, мелкоочаговые кровоизлияния в межальвеолярные перегородки, которые значительно утолщены. Эпителий альвеол с явлениями гиперплазии, клетки округлой формы, ядра их крупные.
Изменения в легких, найденные при микроскопии, способны объяснить тот выраженный альвеолярно-капиллярный барьер для прохождения газов, который так четко проявляется в клинической картине.
В возникновении интерстициального легочного отека немалую роль играет инфекция. Установлено, что интерстициальный отек развивается при заражении обезьян стафилококковым энтеротоксином В, однако у них выявлялись также признаки дегенерации капиллярного эндотелия.
Многочисленные фибробластические пролиферации наблюдаются как в интерстициальной ткани, так и в альвеолярных клетках, но пролиферативные процессы начинают проявляться лишь через несколько дней после начала заболевания и в ряде случаев после начала лечения 100% О2.
При использовании метода ультратонких срезов и электронной микроскопии обнаруживаются изменения во всех клеточных элементах альвеол. Обычно возрастает число мембранозных (I тип) и гранулярных (II тип) выстилающих альвеолярных клеток. Увеличивается также количество макрофагов. Все эти клетки принимают участие в процессах регуляции функции поверхностной активности альвеол и их очищения. Пока неясно, какое отношение к сурфактанту имеют различные типы альвеолярных клеток. Очевидно, что появление в альвеолярной поверхности гранулярных клеток или макрофагов сочетается с уменьшением содержания сурфактанта в легких. Возможно, гранулярные клетки являются поглотителями сурфактанта, количество которого у больных с СДРВ существенно уменьшается.
Лечение. Синдром дыхательных расстройств является, как правило, одним из проявлений критического состояния. В связи с этим лечение его складывается из двух компонентов — общего, осуществляемого в зависимости от характера общей патологии, и специфического, направленного на коррекцию гипоксии. В большинстве случаев это респираторное лечение. Конечно, граница между общим и специфическим лечением весьма условна. Очевидно, например, что степень и выраженность дыхательной недостаточности зависят также от характера инфузионной терапии. Вместе с тем важен принцип комплексности ведения таких больных, в лечении которых имеет значение любое, даже самое мелкое обстоятельство.
Второе положение, которое хотелось бы подчеркнуть — особые условия и длительность лечения. Иногда оно продолжается неделями и происходит в условиях всякого рода психологических давлений на врача, в том числе со стороны родственников больного. Это придает особую окраску всему процессу лечения и требует большого напряжения моральных сил лечащего врача и всего персонала. Нередко благоприятный прогноз лечения полностью отсутствует, и это понимает не только врач, но и персонал отделения. Задача лечащего врача заключается в том чтобы дать правильную установку персоналу на все происходящее и не позволить угаснуть остаткам разумного оптимизма, без которого вообще невозможно выхаживание подобных больных.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 |
