
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
• уточнение характера нарушения (дефи
цит синтеза, специфический или неспеци
фический ингибитор).
2. Контроль проводимой терапии.
Врожденные геморрагические заболевания
Наследственные геморрагические коагулопатии
Наследственные геморрагические коагулопатии, как правило, связаны с генетически детерминированным дефицитом активности факторов свертывания крови. В подавляющем большинстве случаев имеется изолированный дефект одного из факторов, однако возможны комбинированные дефекты. В редчайших случаях геморрагические проявления могут быть связаны со значительным усилением активности антикоагулянтов, например плазминогена, что, как правило, является следствием генетически детерминированного дефицита ингибиторов.
Наследственная недостаточность факторов свертывания встречается в популяции относительно редко. Болезнь чаще всего имеет семейный характер, проявляется в детстве, а больные концентрируются в гематологических центрах. Среди наследственных геморрагических заболеваний наиболее частой является болезнь Виллебранда, которая в некоторых популяциях встречается с частотой до 1%. Однако классически болезнь Виллебранда не относят к коагулопатиям. Среди наследственных геморрагических коагулопатии
наиболее распространены гемофилия А и В, гораздо реже встречается дефицит ф. VII (гипопро-конвертинемия). Дефицит других факторов свертывания встречается еще реже, а его распространенность сильно зависит от особенностей популяции. Например, дефицит ф. ХI широко распространен в популяции евреев-ашкенази, а в других популяциях встречается лишь спорадически. Гомозиготный дефицит ф. Х, приводящий к тяжелым геморрагическим проявлениям, встречается чаще в популяциях, где распространены родственные браки.
Лечение заболеваний, связанных с наследственным дефицитом факторов свертывания, в основном заключается в коррекции дефицита препаратами, содержащими отсутствующий фактор. Такие препараты производят из донорской плазмы или получают генноинженерным путем (рекомбинантные препараты). Также для лечения может применяться свежезамороженная или лиофилизированная плазма или ее первичные производные - криопреципитат или супер-натант.
Патология гемостаза
Гемофилия А
Гемофилия А - геморрагическая коагулопа-тия, связанная со снижением активности фактора VIII в крови. Выделяют наследственную форму гемофилии, связанную с мутациями гена фактора VIII и приобретенную гемофилию.
Наследственная гемофилия А
Ген фактора VIII расположен в Х-хромо-соме, наследственная гемофилия А - заболевание, сцепленное с полом. Тяжелый врожденный дефицит ф. VIII встречается у лиц мужского пола. Женщины являются носителями гена, но у них очень редко возникают геморрагические проявления, связанные с дефицитом ф. VIII (рис. 130). Девочки могут иметь клинические проявления врожденной гемофилии в нескольких случаях: если обе Х-хромосомы с дефектным геном ф. VIII (отец болен гемофилией, а мать является носителем гена гемофилии и передала ей дефектную хромосому); если активна только одна Х-хромосома с дефектным геном ф. VIII (например, при синдроме Шере-шевского-Тернера) и др.
Гемофилия А - наиболее распространенная геморрагическая коагулопатия. Встречается, по разным данным, с частотой от 3 до 20 случаев на 100 000 мужского населения. Примерно в половине случаев диагностируется тяжелая форма за-
болевания. В настоящее время существует несколько классификаций гемофилии по тяжести. Все они основаны на определении активности ф. VIII в крови. В табл. 38 приведена классификация гемофилии А, рекомендованная Всемирной федерацией гемофилии.
Помимо приведенной, предложены другие классификации, в которых тяжелая форма диагностируется при уровне фактора до 2 или 3%.
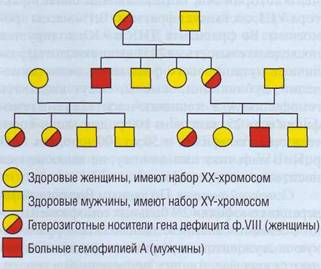
Рис. 130. Генетическое древо наследственной гемофилии. Поскольку ген ф. VIII и ф. IХ находится в Х-хромосоме и отсутствует в Y-хромосоме, риск передачи его от матери составляет 50%, а риск рождения мальчика - 25%
Клиническая классификация гемофилии А
Таблица 38
|

Патология гемостаза
При тяжелой форме гемофилии А активность ф. VIII в крови практически не меняется в течение жизни. При среднетяжелой - возможны колебания в незначительных пределах, а при легкой форме активность фактора может изменяться, в том числе повышаться, особенно при применении синтетических аналогов ва-зопрессина.
Генетика гемофилии А. Врожденная гемофилия А обусловлена дефектами в Х-хромосо-ме, в которой ген, определяющий синтез фактора VIII, составляет фрагмент 0,1% массы хромосомы. Во фрагменте ДНК в 9 Kb кодируется последовательность из 2351 аминокислоты, различные мутации этого фрагмента, включая де-леции, дубликации, замены, могут вызывать гемофилию А. У женщины-носителя гена гемофилии есть 25 шансов из 100 родить дочь-носителя гена гемофилии и 50 из 100 - родить здорового мальчика или девочку, не являющуюся носителем гена гемофилии.
Семейный анамнез. По данным Всемирной федерации гемофилии 2/3 больных гемофилией имеют в семейном анамнезе данные за геморрагическую коагулопатию, а у трети - нарушения выявляются впервые. Анализ, проведенный в гематологическом центре Измайловской ДГКБ г. Москвы, показал другие результаты: среди семей, в которых есть дети, больные гемофилией, лишь около 1/3 до момента рождения больного ребенка знали о наличии геморрагических проявлений у членов семьи.
Анализ ДНК гена фактора VIII в настоящее время является важной частью обследования семьи больного гемофилией. В развитых странах в программы медицинского страхования больных гемофилией введен генетический анализ больных и их семей, что имеет большое практическое значение. Во-первых, это позволяет проводить семейную консультацию с оценкой риска рождения детей с гемофилией у родственников больного. Во-вторых, генетический анализ позволяет провести диагностику гемофилии у плода на ранних сроках беременности. В-третьих, выяснение характера мутации позволяет предсказать риск развития ингибитора к вводимому с лечебными целями фактору VIII. К сожалению, в нашей стране до настоящего времени генетический анализ малодоступен.
Клинические проявления гемофилии А
Для гемофилии А характерны отсроченные кровотечения и кровоизлияния, возникающие после травмы. Поскольку при гемофилии не страдает первичный тромбоцитарный гемостаз, при травмах сосудов небольшого калибра кровотечения останавливаются. Однако аномально длительный процесс свертывания крови не позволяет создать своевременно плотный тромб, что приводит к рецидиву кровотечения. В зависимости от тяжести гемофилии без адекватного лечения вторичное кровотечение может остановиться через некоторое время, но может длиться очень долго, приводя к тяжелой анемии. Очень характерным для гемофилии является кровоизлияние в элементы опорно-двигательного аппарата. Гематомы мышц и рецидивирующие кровоизлияния в суставы приводят этих больных к нарастающему остеопорозу, мышечной дистрофии, артрозам крупных суставов. Значительное поражение опорно-двигательного аппарата у неправильно леченных пациентов с тяжелой гемофилией А формируется к младшему школьному возрасту.
Наиболее значимыми осложнениями гемофилии являются поражение опорно-двигательного аппарата, хроническая постгеморрагическая (железодефицитная) анемия. У пациентов, имевших в анамнезе внутричерепные кровоизлияния, выявляется остаточная неврологическая симптоматика.
Лечение гемофилии
Основным патогенетическим методом лечения гемофилии А является введение препаратов фактора VIII. Поскольку ф. VII содержится в цельной крови и плазме, это были первые препараты, которые использовали для остановки острых кровотечений. Однако в настоящее время от их использования отказались из-за целого ряда серьезных недостатков. Во-первых, относительно низкое содержание ф. VIII в единице объема не позволяет создать в крови больного активность, достаточную для достижения гемостаза в сложных случаях. Во-вторых, поскольку содержание ф. VIII в каждой конкретной дозе неизвестно, нельзя точно дозировать препарат. В-третьих, имеется высокий риск инфицирования гемотрансмиссивными инфекциями (гепатитами и ВИЧ). В-четвертых, имеется проблема
Патология гемостаза
хранения. Цельная кровь и плазма могут храниться только при температурах ниже -20 °С ограниченное время, что исключает их применение вне больницы. В-пятых, при частом применении возрастает число аллергических и анафилактических реакций.
Препаратом следующего поколения стал кри-опреципитат - осажденная при +4 °С часть плазмы, содержащая большое количество ф. VIII, фибриногена и ф. ХIII. По сравнению с кровью и плазмой криопреципитат содержит большее количество ф. VIII на единицу объема, что позволяет останавливать кровотечения даже при некоторых хирургических вмешательствах. Однако в остальном криопреципитат обладает теми же недостатками. В качестве гемостатического препарата при гемофилии А в развитых странах он практически не применяется.
Революционным шагом стала разработка высокоочищенных препаратов ф. VIII. Это позволило создать препараты с высокой активностью ф. VIII в малом объеме, что решило проблему достижения любой необходимой концентрации ф. VIII в крови реципиента. К тому же разработанные методы удаления вирусов позволили свести на нет риск заражения наиболее значимыми инфекциями, передающимися через кровь, - ВИЧ, гепатитами В и С. Современные препараты концентратов ф. VIII представляют собой лиофилизированный порошок с точно определенной активностью ф. VIII. Препарат может храниться при комнатной температуре до 6 месяцев, а при +2... +8 °С - до 2 лет, что позволяет легко использовать его в домашних условиях. Однако такие препараты стоят дорого. Создание концентратов ф. VIII сделало возможным их профилактическое введение для предотвращения кровоизлияний в суставы. В настоящее время разработано четвертое поколение ан-тигемофильных препаратов - концентраты ре-комбинантного ф. VIII. При их производстве вообще не применяются компоненты крови человека, что теоретически полностью исключает риск инфицирования гемотрансмиссивными вирусами. К сожалению, стоимость этих препаратов значительно выше, чем препаратов производных человеческой плазмы.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 |
