
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
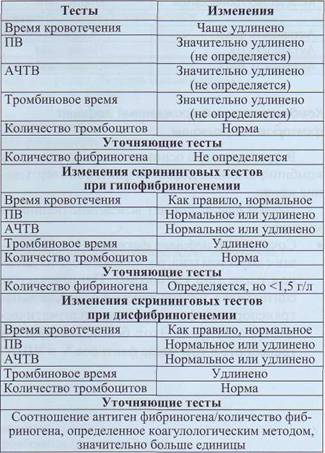
О состоянии гипофибриногенемии говорят в том случае, если содержание фибриногена в плазме менее 1 г/л. Клинические проявления аналогичны проявлениям при афибриногенемии, однако менее выражены.
Диагноз дисфибриногенемий соответствует состоянию, при котором изменена структура фибриногена, однако содержание самого белка в крови (антигена) нормальное или снижено непропорционально функции. Дисфибриногенемий могут проявляться кровотечениями, тромбозами или не иметь никаких проявлений. Клинические проявления геморрагических дисфибриногенемий сходны с проявлениями гипофибриногенемии.
Лабораторная диагностика количественных и качественных нарушений фибриногена основана на изменении стандартных тестов коагулограммы (табл. 47). Для установления диагноза дисфибриногенемий показано проведение дополнительных тестов. Часто этот диагноз можно поставить только после исследования гена ф. I.
Таблица 47
Изменение скрининговых тестов при афибриногенемии
Патология гемостаза
Дополнительным тестом, позволяющим по косвенным признакам заподозрить дисфибрино-генемию, является тромбоэластография.
Дефицит факторов контактной активации
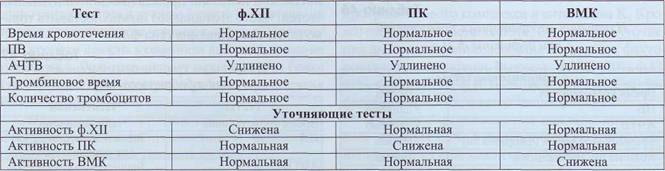
Дефицит ф. ХII, прекалликреина (ПК) и высокомолекулярного кининогена (ВМК) нельзя в полной мере отнести к геморрагическим заболеваниям. Дефицит активности ВМК или ПК клинически никак не проявляется.
У пациентов с дефицитом ф. ХII (болезнь Ха-гемана) имеются разнонаправленные тенденции. У большинства из них, даже при глубоком дефи-
ците, нет геморрагических проявлении, однако у некоторых пациентов этой группы имеет место повышенная кровоточивость. Некоторые пациенты с дефицитом ф. ХII имеют тенденцию к тром-ботическим проявлениям.
Распространенность дефицита ф. ХII в популяции довольно высока. Большинство случаев удлинения АЧТВ у пациентов без клинических проявлений связано с этой патологией. По некоторым данным частота гетеро - и гомозиготных форм дефицита ф. ХII в популяции достигает 1,5-3%.
Лабораторные данные при дефиците ф. ХII, ПК, ВМК представлены в табл. 48.
Таблица 48
Изменения скрининговых тестов при дефиците факторов контактной активации
|
Комбинированный врожденный дефицит факторов свертывания
Встречаются два основных типа врожденных комбинированных дефектов факторов свертывания крови.
Первый тип возникает вследствие общности мутации:
• Сочетанный дефицит факторов V и VIII связан с дефектом гена, расположенного на длинном плече 18-й хромосомы. Ген отвечает за синтез белка, участвующего в осуществлении транспортной функции в эндоплазматичес-ком ретикулуме. Этот белок участвует в транспорте в том числе факторов V и VIII.
• Комбинированный дефицит факторов II, V,
IX, X возникает у пациентов с 1-м доминант
ным типом эластической псевдоксантомы,
при варфариновой эмбриопатии, мутации
гена гаммаглутамилкарбоксилазы.
• Комбинированный дефицит факторов VIII и IX,
гены которых расположены на Х-хромосоме,
возникает вследствие дефекта хромосомы,
затрагивающего оба гена.
• Комбинированный дефицит факторов VII и X,
связанный с делецией 13-й хромосомы.
Второй тип возникает вследствие независи
мых мутаций гена у одного пациента.
Диагностика комбинированных мутаций проводится по стандартному плану.
Патология гемостаза
Врожденные нарушения функции тромбоцитов
Врожденные нарушения функции тромбоцитов - достаточно гетерогенная группа тромбоци-топатий. До настоящего времени исследуются внутриклеточные механизмы активации и функционирования тромбоцитов, которые обеспечиваются разнообразными функциональными механизмами. Поэтому существует несколько предложений по классификации наследственных тром-боцитопатий, одна из них, которая учитывает наиболее разработанные представления о метаболизме и регуляции тромбоцитарных функций, представлена в табл. 49.
На рис. 131 показана схема нарушения функции тромбоцитов при наиболее распространенных врожденных дефектах.
Клинические проявления врожденных нарушений функции тромбоцитов для большинства заболеваний сходны. Отмечается разной
степени выраженности кровоточивость по микроцирку ляторному типу: петехии, экхимо-зы, длительные первичные кровотечения после травм слизистых, первичные послеоперационные кровотечения, носовые кровотечения, маточные кровотечения на фоне менструации. В большинстве случаев геморрагический синдром выражен не сильно и редко угрожает жизни. Исключение составляют такие заболевания, как тромбастения Гланцмана и синдром Берна-ра-Сулье, при которых возможны опасные для жизни проявления: внутричерепные кровоизлияния, тяжелые маточные и носовые кровотечения, кровотечения со слизистых других локализаций, послеоперационные кровотечения. При тромбастении Гланцмана описаны гемартрозы с развитием артропатии, сходной с гемо-филической.
Классификация врожденных нарушений функции тромбоцитов (Rao. Am J Med Sci 1998; 316: 69-77)
Таблица 49
|

Патология гемостаза
Изменения лабораторных тестов при врожденных нарушениях функции тромбоцитов представлены в табл. 50.
Индуцированная агрегация на различные активаторы является одним из наиболее показательных лабораторных тестов для выявления на-
следственных нарушений функции тромбоцитов (рис. 132). Наиболее значимые лабораторные тесты, используемые для диагностики врожденных нарушений функций тромбоцитов, суммированы в табл. 51.
|
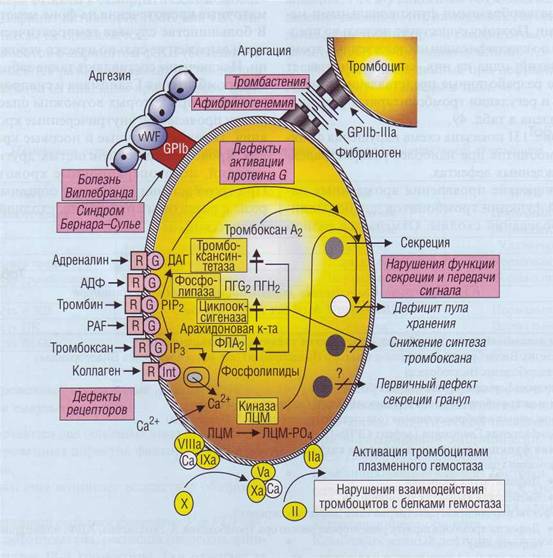
Рис. 131. Схема механизмов развития врожденных нарушений функции тромбоцитов. ДАГ - диацилглицерол, ЛЦМ - легкие цепи миозина, ПГС2 и ПГН2- эндоперекиси, ФЛА2- фосфолипаза А2, G - G-белок, 1Р3 - инозитолтрифосфат, PAF - тромбоцит-активирующий фактор, Р1Р2 - фосфатидилинозитол-4,5-бисфосфат, R - рецептор, vWF - фактор Виллебранда
Патология гемостаза
Врожденные нарушения функции тромбоцитов
Таблица 50
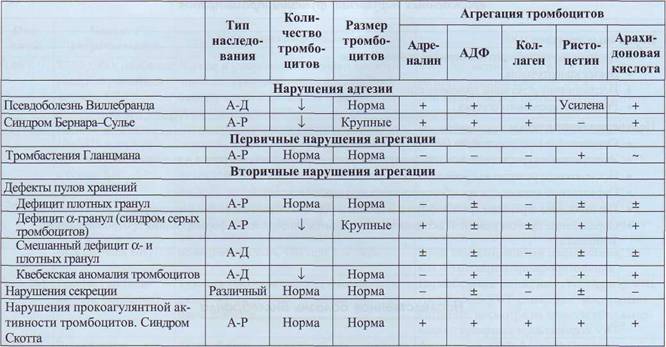
А-Р - аутосомно-рецессивный, А-Д - аутосомно-доминантный, «+» - присутствует, нормальная, «-» - отсутствует или снижена, «±» - вариабельна либо слегка снижена.
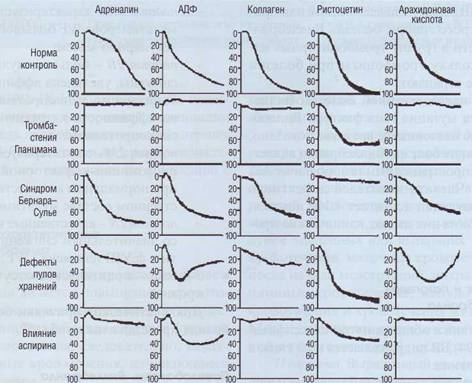
Рис. 132. Агрегация тромбоцитов с различными индукторами у здоровых людей и при врожденных нарушениях функции тромбоцитов
Патология гемостаза
Наиболее значимые лабораторные тесты, используемые для диагностики врожденных нарушений функций тромбоцитов
Таблица 51

|

Наследственная болезнь Виллебранда

Болезнь Виллебранда (БВ) - геморрагическое заболевание, являющееся следствием качественных или количественных нарушений фактора Виллебранда. БВ бывает наследственной или приобретенной. Строго говоря, болезнь Виллебранда нельзя отнести в группу тромбоцитарных нарушений, поскольку тромбоциты при болезни Виллебранда не страдают.
Причиной наследственной болезни Виллебранда является мутация гена фактора Виллебранда (vWF). К настоящему времени показано, что наследственная болезнь Виллебранда является наиболее распространенным геморрагическим заболеванием. Частота носителей дефектного гена vWF в популяции достигает 1:100 человек, но лишь 10-30% из них имеют клинические проявления.
Классификация и патогенез болезни Виллебранда
В соответствии с общепринятой классификацией (Sadler, 1994) БВ подразделяется на 3 типа, а тип 2 - на 4 подтипа:
• 1-й тип - наследственное заболевание с частичным дефицитом vWF в крови и нормальным распределением мультимеров vWF;
• 2-й тип - наследственная патология с каче
ственным изменением vWF. Второй тип под
разделяют на 4 подтипа:
подтип 2А - характеризуется снижением мультимеров vWF большой и средней молекулярной массы;
- подтип 2В - большие мультимеры vWF
снижены, увеличена аффинность к тром-
боцитарному гликопротеину 1Ь и сниже
на аффинность к другим рецепторным
гликопротеинам;
- подтип 2М - характеризуется сниженной
ристомицин-кофакторной активностью
при нормальном количественном и каче
ственном составе мультимеров vWF;
- подтип 2N - качественные варианты vWF
со значительным снижением аффиннос
ти к ф. VIII (снижена vWF:FVIIIB);
• 3-й тип - практическое отсутствие vWF в
крови.
Диагностические признаки болезни Виллебранда представлены в табл. 52.
Псевдоболезнь Виллебранда
Псевдоболезнь Виллебранда (тромбоцитарный тип) возникает вследствие повышенного связы-

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 |
