
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
При неонатальной аллоиммунной тромбо-цитопении антитела вырабатываются в результате иммунизации матери аллоантигенными де-
терминантами, содержащимися на тромбоцитах отца и ребенка. Тромбоцитопения в этом случае сохраняется у новорожденного в течение 2-
3 недель.
Гаптеновые (гетероиммунные) тромбоцитопении
Гаптеновые тромбоцитопении сопровождаются выработкой антител против измененных или чужеродных структур на поверхности тромбоцитов, появляющихся в результате воздействия некоторых лекарственных препаратов (табл. 58). Хинин и препараты хининового ряда способны стимулировать образование гаптено-вых антител, так как они связываются с тромбо-цитарными рецепторами с образованием комплексов.
Следует подчеркнуть, что какой бы ни была причина нарушения функции тромбоцитов, при тромбоцитопении следует избегать лекарственных средств, способных нарушать эти функции, в частности, следует отказаться от аспирина и некоторых других нестероидных противовоспалительных средств.
Тромбоцитопения, вызванная гепарином
Тромбоцитопения развивается примерно у 5% больных, получавших бычий гепарин, и 1% больных, получавших свиной гепарин. У больных с гепариновой тромбоцитопенией прогрессивно увеличивается риск тромбоза, возникают угрожа-
Препараты, способные вызвать лекарственную тромбоцитопению
Таблица 58
|

Патология гемостаза
ющие жизни артериальные тромбы (рикошетные тромбозы). Патогенез гепарин-индуцированной тромбоцитопении (ГИТ) связан с действием патогенных гепарин-зависимых IgG-антител (ГИТ-IgG). Тромбоцитарный фактор 4 является гепа-рин-связывающим белком. На поверхности тромбоцитов формируется мультимолекулярный комплекс между IgG и гепарином/тромбоцитарным фактором 4 (рис. 135). Мультимолекулярный комплекс связывается со специфическим рецептором (FcγRIIA) на тромбоцитарной мембране. Предрасположенность к этому осложнению связана с мутацией в FcγRIIA-гене. В результате в молекуле FcyRIIA-рецептора происходит замена Arg 131 —>His 131, и пациенты с такой мутацией становятся склонны к развитию гепарин-индуци-рованных рикошетных тромбозов. В будущем, по-видимому, молекулярно-генетическая диагностика позволит идентифицировать пациентов с повышенным риском развития гепариновой тромбоцитопении и рикошетных гепариновых тромбозов. В настоящее время диагностика таких состояний проводится сочетанием метода ELISA с использованием антител против комплекса гепарин - тромбоцитарный фактор 4 или методом определения агрегации или освобождения 14С-се-ротонина из предварительно нагруженных тромбоцитов под действием гепарина. Однако эти методы неспецифичны и часто бывают нечувствительными.
Покрытые IgG тромбоциты активно удаляются из системы циркуляции макрофагами. ГИТ-IgG способны повреждать эндотелиальные клетки. Это связано с тем, что гепарансульфат гликокаликса эндотелия как структурный аналог гепарина может вступать в качестве антигена во взаимодействие с ГИТ-IgG. Затем возможно развитие иммунных реакций на поверхности эндотелия, адгезия в этих зонах макрофагов и развитие пристеночного тромба.
Следует отметить, что низкомолекулярные гепарины (фраксипарин, фрагмин, ловенокс, клексан) не вызывают тромбоцитопении, вероятно, из-за меньших, чем требуется для образования иммунного комплекса, размеров молекул этих препаратов.
Если у больного, получавшего гепарин, развивается тромбоцитопения, необходимо провести тест для определения гепарин-ассоциирован-ной агрегации тромбоцитов и гепарин-индуцированной тромбоцитопении. Для этой цели с успехом использовали импедансный и люминесцентный агрегометр «Chrono-log» (рис. 77), который одновременно с агрегацией тромбоцитов в цельной крови позволяет определять высвобождение из гранул АТФ как конечную точку активации тромбоцитов, вызванной антителами. Методика позволяет в течение 1 часа поставить диагноз нарушения функции тромбоцитов и дефектов накопления.
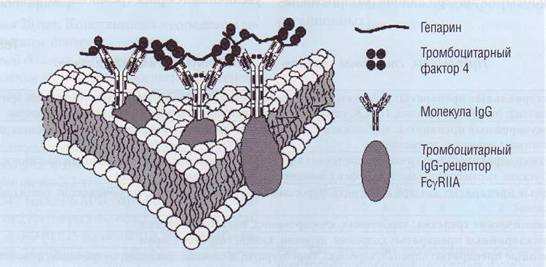
Рис. 135. Активация тромбоцитов гепарином за счет образования на поверхности мембран мультимолекулярного комплекса между гепарин-зависимыми lgG-антителами и тромбоцитарным фактором 4
Патология гемостаза
|
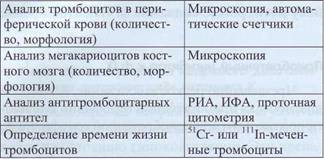
Дифференциальная диагностика тромбоцито-пений должна быть направлена на выяснение природы тромбоцитопении: 1) иммунной природы (иммунные тромбоцитопении), 2) тромбоцитопении, возникающей в результате угнетения тром-боцитопоэза в костном мозге, или 3) наследственных тромбоцитопении, ассоциированных с тром-боцитопатиями. Лабораторные методы, применяемые при диагностике тромбоцитопении, представлены в табл. 59.
Таблица 59
Методы лабораторной диагностики тромбоцитопении
Приобретенный дефицит факторов свертывания крови
 Развитие специфического ингибитора
Развитие специфического ингибитора
Чаще всего встречается развитие ингибитора к факторам VIII и IX у пациентов, страдающих соответственно гемофилией А и В и получающих специфическую заместительную терапию. Однако описаны приобретенные формы гемофилии А, болезни Виллебранда, реже приобретенный дефицит других факторов свертывания у лиц, не страдавших врожденными нарушениями.
Основными предрасполагающими факторами являются:
• аутоиммунные заболевания;
• онкологические заболевания;
• перенесенная инфекция;
• прием некоторых лекарственных препаратов.
Возможно возникновение антител без явных
провоцирующих воздействий.
Приобретенный ингибитор к фактору VIII (приобретенная гемофилия А)
Возникает вследствие образования антител к собственному ф. VIII. Чаще развивается у лиц старше 50 лет, однако описаны случаи заболевания детей. Мужчины и женщины страдают оди-
наково часто. Расчетная частота развития ингибитора к ф. VIII у пациентов, не болевших гемофилией, составляет 1 случай на миллион человек в год.
У описанных в литературе за последние 10 лет 215 пациентов в 50% случаев ингибитор развился спонтанно. В качестве фоновых заболеваний называют: системные заболевания соединительной ткани, ревматоидный артрит, многоформную эритему, герпетиформный дерматит, аллергические реакции на пенициллин, бронхиальную астму, неспецифический язвенный колит, болезнь Крона, терапию α-интерфероном, моноклональные гам-мапатии, беременность и послеродовый период.
У части пациентов ингибитор спонтанно исчез через 12-18 месяцев после развития, однако остальным потребовалось специальное лечение. Терапия аналогична лечению ингибиторной формы гемофилии А.
Диагностика основана на клинических (признаки развившегося геморрагического заболевания с кровоточивостью по гематомному типу) и лабораторных данных (снижение активности ф. VIII, наличие специфического ингибитора к ф. VIII).
Клинический пример 8
Больная 68 лет. Поступила в отделение гастроэнтерологии. На 3-й день в области бедер, ягодицы появились обширные гематомы. Переведе-
на в гематологическое отделение с диагнозом: геморрагический синдром неясной этиологии.
Лабораторный анализ: АЧТВ 161 с (норма 35-45 с), ПТ 93%, ТВ 34 с (норма 28-30 с), фибриноген 2,4 г/л, лизис эуглобулиновой фракции 140 мин,
Патология гемостаза
ретракция кровяного сгустка 46%, фактор VIII 1,3%, агрегация тромбоцитов с АДФ 72%.
Заключение: значительное удлинение времени свертывания крови при активации по внутреннему пути, резкое снижение ф. VIII. Подозрение
на ингибиторную форму гемофилии А. Для подтверждения необходимо определение ф. VIII:Ag. Больная направлена в гематологический центр, где была подтверждена приобретенная (ин-гибиторная) гемофилия А.
Приобретенный ингибитор к ф./Х
Чрезвычайно редкое заболевание. Клиника и диагностика приобретенной гемофилии В аналогичны таковым при приобретенной гемофилии А.
Ингибитор к фактору Вимебранда (приобретенная болезнь Виллебранда, приобретенный синдром Виллебранда)
Приобретенная болезнь Виллебранда (приобретенный синдром) лабораторно подобна нарушениям, характерным для врожденной болезни Виллебранда. Редкое заболевание. Возникает как спонтанно, так и на фоне ряда заболеваний: патология сердца и сосудов, онкологические заболевания, системные заболевания соединительной ткани, гипотиреоз, опухоль Вильмса, прием цип-рофлоксацина и др. Патогенетические механизмы формирования приобретенного синдрома Виллебранда:
• Специфические антитела к ф. VIII/vWF.
• Неспецифические антитела, которые форми
руют иммунные комплексы и приводят к бо
лее активному клиренсу vWF.
• Абсорбция vWF клетками злокачественных
опухолей.
• Повышение протеолитической деградации
vWF.
• Потеря тяжелых молекул vWF в условиях
стресса, связанного с высоким напряжением
сдвига при активном кровотоке.
• Снижение синтеза или высвобождения vWF.
Диагностика: выявление развившихся при
знаков приобретенного геморрагического забо
левания, сходных с наследственной болезнью
Виллебранда. Лабораторная диагностика приоб
ретенной болезни Виллебранда аналогична диаг
ностике врожденной болезни Виллебранда. При
этом, помимо состояния гемостаза, необходимо
выявить фоновые заболевания.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 |
