
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Key words: aplastic anemia, chromosomal abnormalities, children.
Вступ. Апластична анемія (АА) розвивається внаслідок зупинки проліферації та загибелі стовбурових кровотворних клітин (СКК). АА характеризується практично повною зупинкою кровотворення, що морфологічно проявляється в заміщенні функціонально активного кісткового мозку інертною жировою тканиною з одночасною відсутністю аномальної інфільтрації і без збільшення ретикулуму та панцитопенією у периферичній крові (ПК) [1].
До числа можливих причин виникнення АА сучасна наука включає: а) аномалії СКК, б) аномалії гемопоетичного мікрооточення, в) імунологічно-опосередковані зміни. Наявність аномалій хромосом у СКК свідчить про високий ризик розвитку мієлодиспластичних синдромів (МДС), гострої мієлоїдної лейкемії (ГМЛ) чи нічної пароксизмальної гемоглобінурії. Таким чином, розвивається гіпотеза, що АА викликана аномальним клоном. Тим більше, що клінічні риси АА та МДС можуть перекриватися, і типова МДС може проявлятися як ускладнена АА [2].
Ззалежно від ступеня периферичної цитопенії (анемія, гранулоцитопенія, тромбоцитопенія, ретикулоцитопенія) виділяють наступні типи АА – дуже тяжку, тяжку форму та середньої тяжкості. Серед клональних аномалій хромосом у дітей найчастіше описана моносомія Хр 7 (-7) [3]. Наступною за частотою виявляється трисомія Хр 8 (+8) [4]. Крім того, описані випадки АА з жировим заміщенням у КМ та t(9;22)(q34;q11) [5].
Метою дослідження був аналіз каріотипу клітин КМ у дітей із цитопенічним синромом на етапі встановлення діагнозу АА.
Матеріали та методи дослідження. Цитогенетичне дослідження клітин КМ на час встановлення діагнозу проведено у 42 пацієнтів, що обстежувалися з приводу цитопенічного синдрому. Серед них було 15 дівчаток та 27 хлопчиків, середній вік – 11,2 роки (від 1,9 до 18 років).
Середній вміст лейкоцитів у групі становив 3,2´109/л (від 1,1 до 5,2´109/л), гемоглобіну – 75 г/л (від 48 до 114 г/л), тромбоцитів – 82,3´109/л (від 1 до 116´109/л). За даними трепанобіопсії КМ, у всіх цих пацієнтів відмічалося зниження клітинності КМ до резидуальних 25,0–30,0% із переважаючим заміщенням кровотворного КМ на жировий. У мієлограмах реєстрували аплазію різного ступеня гранулоцито - та мегакаріоцитопоезу, у 19 випадках – з ознаками дизеритропоезу (мегалобластоз, дисоціація дозрівання ядра та цитоплазми еритрокаріоцитів, наявність двоядерних форм, підвищений вміст «між’ядерних та міжцитоплазматичних мостиків», аномальні форми нормоцитів). Тобто, гістологічними та морфологічними методами було підтверджено діагноз АА.
Всі пацієнти з АА були поділені на дві групи. До групи з АА середньої тяжкості віднесено 26 пацієнтів. Серед них було 10 дівчаток та 16 хлопчиків. Середній вік у групі становив 11,3 років. Рівень гранулоцитів на час обстеження у цій групі був менший за 1000 гранулоцитів в мкл. До другої групи ввійшли пацієнти з дуже тяжкою формою АА – 4 пацієнти (рівень гранулоцитів – менше 200 гранулоцитів в мкл) та з тяжкою формою АА – 12 пацієнтів (рівень гранулоцитів – менше 500 гранулоцитів в мкл).
Для цитогенетичних досліджень препарати метафазних хромосом готували за загальновизнаною методикою [6] і фарбували за GTG-методом. Виявлені хромосомні аномалії описували згідно з міжнародною номенклатурою ISCN 2009 [7].
Результати дослідження та їх обговорення. У результаті цитогенетичних досліджень каріотипи клітин КМ пацієнтів з АА за структурою клонів розподілили таким чином:
- нормальний (Н) – 19,0% випадків,
- нормальний та білятетраплоїдний клони (Н/4n±) – 35,7%,
- аномальний та нормальний (A/Н) – 11,9%,
- аномальний, білятетраплоїдний та нормальний (A/4n±/Н) – 19,0%,
- еволюція клону (Е) – 9,5%,
- незалежні клони (НК) – 4,8%.
Представлені результаті свідчать, що частота нормального клону у поєднанні з мозаїчним каріотипом (нормальний та білятетраплоїдний клони) реєструвалася більш ніж у половині випадків (54,7%). Частота мозаїчних каріотипів становила 81,0%, частота аномальних каріотипів – 45,2%, що перевищує результати інших досліджень у 2-4 рази [4].
Аналіз кількісних та структурних аномалій хромосом відносно плоїдності представлено в табл. 1. У третині випадків зареєстровані псевдодиплоїдні клони, які формувалися за рахунок незбалансованих або збалансованих та незбалансованих аномалій хромосом. У всіх випадках з аномальними клонами були присутні і нормальні клони, що може свідчити про збереження цитогенетично нормальних каріотипів.
Таблиця 1
Розподіл кількісних аномалій хромосом відносно плоїдності в клітинах кісткового мозку при апластичній анемії у дитячому віці
Рівень плоїдності | Абсолютна кількість спостережень | Відсотковий вміст,% |
Диплоїдний | 23 | 54,7 |
Псевдодиплоїдний | 14 | 33,3 |
Гіподиплоїдний | 3 | 7,2 |
Гіпердиплоїдний (47–50 хромосом) | 2 | 4,8 |
Додатковий білятетраплоїдний | 21 | 50,0 |
Аналіз структурних перебудов хромосом (Хр) показав, що за типами перебудов реєстрували делецію, транслокацію та ізохромосому, парацентричну інверсію та дуплікацію (рис. 1). Серед цих перебудов домінували делеції – 43,0%. До структурних перебудов залучалися тринадцять хромосом – 3, 4, 6, 7, 9, 11, 12, 13, 14, 16, 17, 21 та 22. Найчастіше це відбувалося у Хр 9 по дисках 9q22, 9q34 (11,9%) і серед них виявлена транслокація t(9;22)(q34;q11), яка характерна для хронічної мієлоїдної лейкемії. Делеції довгих плеч Хр 11, 16, 17 та короткого плеча Хр 17 описані при гострих лейкеміях [8, 9].


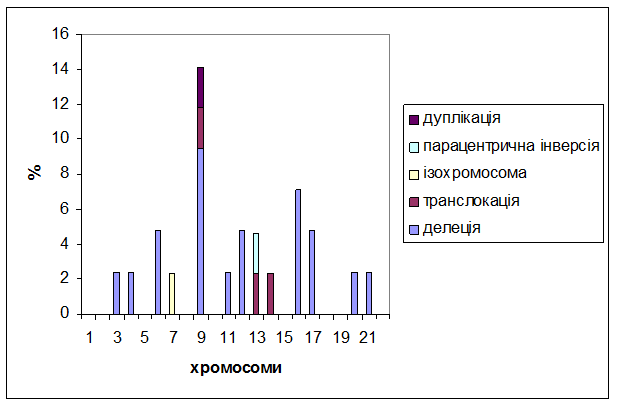
Рис. 1. – Відсотковий розподіл типів структурних перебудов у клітинах кісткового мозку при апластичній анемії у дитячому віці
Таким чином, клітини КМ у пацієнтів з АА дитячого віку представлені широким спектром клонів, моносомією та трисомією хромосом, делецією, транслокацєю, в тому числі t(9;22), парацентричною інверсією, дуплікацією та високою частотою структурних перебудов Хр 9. Привертає увагу наявність складних клональних хромосомних аномалій – еволюції та незалежні клони (9,5% та 4,8% відповідно).
Розподіл каріотипів клітин КМ при АА середньої тяжкості за структурою клонів представлено у табл. 2.
Таблиця 2
Розподіл каріотипів за структурою клонів у клітинах кісткового мозку при апластичній анемії (середньої тяжкості та тяжка форма) у дитячому віці
Каріотип | АА середньої тяжкості | АА тяжка форма |
% | ||
Нормальний | 26,9 | 6,3 |
Нормальний та білятетраплоїдний клони | 30,8 | 43,7 |
Аномальний та нормальний | 19,2 | 0 |
Аномальний, білятетраплоїдний та нормальний | 15,4 | 25,0 |
Еволюція клону | 3,8 | 18,7 |
Незалежні клони | 3,8 | 6,3 |
Як видно з представлених результатів, для обох груп характерний широкий діапазон каріотипів за структурою клонів. Нормальний каріотип як самостійний клон, так і у вигляді мозаїчного каріотипу (нормальний та білятетраплоїдний клон) у обох групах з АА виявлявся у більше, ніж половині випадків – 57,7% та 50,0%, відповідно, що значно нижче, ніж у літературних посиланнях (70,0–90,0%) [4]. Випадки з клональними аномаліями хромосом складали 42,3% та 50,0% відповідно. При цьому частота каріотипів з еволюцією клональних аномалій хромосом була майже у п’ять разів вище у групі з тяжкою формою АА (18,7% проти 3,8%). Найчастіше мозаїчні каріотипи фіксувалися при АА тяжкої форми, ніж при АА середньої тяжкості (93,7% та 73,1% відповідно). Аналіз кількісних аномалій хромосом відносно плоїдності представлено в табл. 3.
Таблиця 3
Розподіл кількісних аномалій каріотипу клітин кісткового мозку при апластичній анемії середньої тяжкості та тяжкій формі у дитячому віці
Рівень плоїдності | АА середньої тяжкості | АА тяжка форма |
% | ||
Диплоїдний | 50,0 | 62,5 |
Псевдодиплоїдний | 34,6 | 31,3 |
Гіподиплоїдний | 11,5 | 0 |
Гіпердиплоїдний (47-50 хромосом) | 3,8 | 6,3 |
Додатковий білятетраплоїдний | 42,3 | 62,5 |
Серед типів перебудов реєстрували втрату генетичного матеріалу (делеція), транслокацію та ізохромосому, при цьому домінувала делеція (18 випадків – 43,0%). До структурних перебудов залучалися одинадцять хромосом. Найчастіше це спостерігалося для Хр 9 (3 випадки – 11,5%), Хр 7 та 16 (по 2 випадки – 7,6%). Делеція довгого плеча Хр 7 більш характерна для МДС [4], аномалії по дискам 12р12 та 16q22 часто зустрічаються при ГМЛ [10].
У групі тяжкої форми АА серед кількісних аномалій хромосом зареєстровані тільки трисомії Хр 11 та маркерна хромосома. У цій групі реєстрували ширший спектр типів перебудов, ніж при АА середньої тяжкості, а саме: делеція, транслокація, дуплікація та парацентрична інверсія. Найчастіше, як і у випадках АА середньої тяжкості, реєстрували делеції – 6 випадків (37,5%). До структурних перебудов залучалися десять хромосом. Найчастіше це була Хр 9 (3 випадки та 4 події – 18,8%, по дисках 9q33, 9q34). У двох випадках реєстрували транслокацію t(9;22)(q34;q11), яка характерна для ХМЛ і зустрічається при ГЛ [6]. Одна з них була виявлена у вигляді варіантної транслокації t(9;22;12)(q34;q11;p11), інша – під час еволюції аномального клону. Дисбаланс геному гемопоетичних клітин, який утворився внаслідок кількісних та структурних перебудов, реєструвався майже у два рази частіше у групі АА середньої тяжкості та становив 62,4% проти 37,5% у групі АА тяжкої форми.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 |
