
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
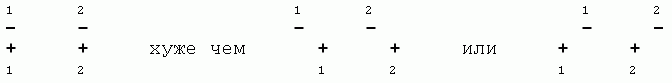
Глядя на схему, нужно помнить, что электрическая энергия растет с уменьшением расстояния r как 1/r. В результате смещения электронов взаимодействия электрон-электрон и ядро-ядро не меняются, но (рассмотрим самый правый рисунок) усиливается притяжение электрона 1 к ядру 2 — причем оно усиливается больше, чем ослабевает притяжение электрона 2 к ядру 1. Поэтому согласованное колебание электронов (а это и есть квантовый эффект) понижает энергию системы. При отдалении атомов их взаимодействие ослабляется. В результате энергия взаимодействия спадает с расстоянием r между ядрами атомов — можно показать, что она спадает пропорционально (1/r)6.
Общая энергия взаимодействия атомов — она еще называется энергией Вандерваальсова взаимодействия — охватывается рисунком 3-1 и приближенно описывается формой Леннард-Джонса:
ULD(r) = E0[(r0/r)12 - (r0/r)6] | (3.1) |
Здесь r0 — как легко проверить, взяв производную ULD по расстоянию r — есть то расстояние, на котором энергия ULD проходит через минимум, а - E0 — глубина этого минимума. Последний член формулы (3.1) — тот, что падает с расстоянием, как (const/расстояние)_в_шестой_степени — соответствует притяжению (знак "минус" перед ним указывет, что соответствующая энергия падает при сближении); а тот, где значится двенадцатая степень, соответствует отталкиванию (этот член положителен, т. е. соответствующая ему энергия растет при сближении).
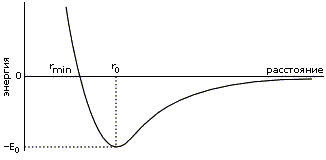
Рис.3-1. Характерный вид потенциала Вандерваальсова взаимодействия.
Формула 3.1 точно описывает только притяжение на больших (когда r << r0) расстояниях. Отталкивание на малых расстояниях она описывает только качественно, как "очень сильное отталкивание, превосходящее любое притяжение, когда r стремится к нулю". То, что формула (3.1) — приближенная, видно хотя бы по тому, что она подразумевает округлость атомов (т. к. ею описываемое взаимодействие не зависит от направления), — а электронное облако у атома, вообще говоря, не круглое, — вспомните хотя бы о торчащих p-электронах. Вообще, правильно и совсем корректно взаимодействие атомов описывает только квантовая механика — но точно рассчитать она может только очень простые системы, вроде атома Не, иона Н2+ или молекулы Н2. Для всего остального приходится брать приближенные, "полуэмпирические" формулы — вроде (3.1) — где вид формулы берется из качественных физических соображений, а параметры (у нас — E0 и r0) — подбираются из опыта. Некоторые основные данные сведены в Таблицу 3/1.
Таблица 3/1. Характерные параметры потенциалов Вандерваальсовых взаимодействий
Взаимодействие | E0, ккал/моль | r0, | rmin, | Вандерваальсов радиус атома, | |
H . . . . H | 0.12 | 2.4 | 2.0 | H: | 1.0 |
H . . . . C | 0.11 | 2.9 | 2.4 | ||
C . . . . C | 0.12 | 3.4 | 3.0 | C: | 1.5 |
O . . . . O | 0.23 | 3.0 | 2.7 | O: | 1.35 |
N . . . . N | 0.20 | 3.1 | 2.7 | N: | 1.35 |
CH2 . . . CH2 | »0.5 | »4.0 | »3.0 | CH2: | »1.5 |
Примечания. Величины E0, r0 для межатомных взаимодействий взяты из R. A.Scott, H. A.Scheraga, J. Chem. Phys. (1965) 45:2091, а величины rmin — из G. N.Ramachandran, V. Sasisekharan, Adv. Prot. Chem. (1968) 23:283. Исходя из этих величин, оценены параметры взаимодействия CH2...CH2. Взаимодействие CH2...CH2 зависит от взаимной ориентации этих групп; поэтому приведенные в таблице числа — приблизительные; их, однако, часто приходится использовать, так как рентген "не видит" водородных атомов в белках.
Обратите также внимание на то, что в таблице дано не только оптимальное расстояние r0, но и rmin — минимальное расстояние, на котором соответствующие атомы встречаются в кристаллах. rmin примерно соответствует той точке Рис.3-1, где энергия при сильном сближении проходит через 0.
Расстояния (r0 , rmin) и энергии (E0) в основном берутся из кристаллов — из их структур (расстояния) и теплот сублимации (энергии). Но кристаллы обычно состоят не из атомов, а из молекул. Например: СН4, С2Н6,... Так что при расчете энергии всегда возникают вопросы типа: каков вклад С...С, каков — Н...Н, каков — С...Н взаимодействий? Разные авторы решают его по-разному — поэтому потенциалы у них порой заметно расходятся. Надо помнить, что нельзя самодеятельно брать одни параметры (например, энергию С...С взаимодействия) у одного автора, а другие — у другого: тут действует принцип "все или ничего", иначе будут ошибки.
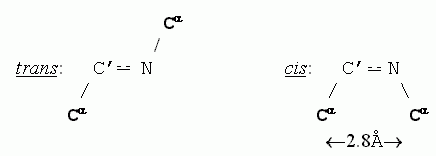
Приведенные в таблице величины помогают понять, почему trans-конформация С'![]() N связи допустима, а ее cis-конформация — нет (для всех аминокислотных остатков, кроме пролина, о чем мы уже говорили): в trans-конформации С'
N связи допустима, а ее cis-конформация — нет (для всех аминокислотных остатков, кроме пролина, о чем мы уже говорили): в trans-конформации С'![]() N связи расстояние между Сaатомами равно 3.8
N связи расстояние между Сaатомами равно 3.8![]() , а в cis - (когда эти атомы наиболее сближены) — всего 2.8
, а в cis - (когда эти атомы наиболее сближены) — всего 2.8![]() , что меньше минимально допустимого расстояния rMIN, равного 3.0
, что меньше минимально допустимого расстояния rMIN, равного 3.0![]() для пары С...С.
для пары С...С.
Из-за жесткости trans-формы С'![]() N связи и из-за того, что в ней Ca-атомы соседних по цепи аминокислот довольно далеко отстоят друг от друга, соседние по цепи остатки могут менять свою конформацию почти независимо друг от друга. Но внутри остатка вращения по углам f и y взаимосвязаны. Изображенные в координатах (f, y) "разрешенные" и "запрещенные" конформации остатка называются картами Рамачандрана (или, более корректно, — картами Рамачандрана-Сасисекхарана).
N связи и из-за того, что в ней Ca-атомы соседних по цепи аминокислот довольно далеко отстоят друг от друга, соседние по цепи остатки могут менять свою конформацию почти независимо друг от друга. Но внутри остатка вращения по углам f и y взаимосвязаны. Изображенные в координатах (f, y) "разрешенные" и "запрещенные" конформации остатка называются картами Рамачандрана (или, более корректно, — картами Рамачандрана-Сасисекхарана).
Прежде чем рисовать их, — посмотрим, какие конформации разрешены (и какие — нет) при вращении по углу f (вокруг N-Ca связи) и по углу y (вокруг Ca-С' связи) в отдельности.
Мы уже знаем, что вращение вокруг этих связей (соединяющих sp3-гибридизованный Сa-атом с sp2-гибридизованными N или C') практически свободно. Однако в cis-конформациях (при f=0o или y=0o) вращающиеся вокруг таких связей атомы (C'i-1 и C'i при вращении по углу f вокруг N-Ca связи, и атомы Ni и Ni+1 при вращении по углу y вокруг Ca-C' связи, см. Рис.3-2; i-1, i, i+1 - номера последовательных остатков в цепи) сближаются особенно сильно, и здесь возможен запрет конформации из-за отталкивания этих атомов — как говорят, стерический запрет.
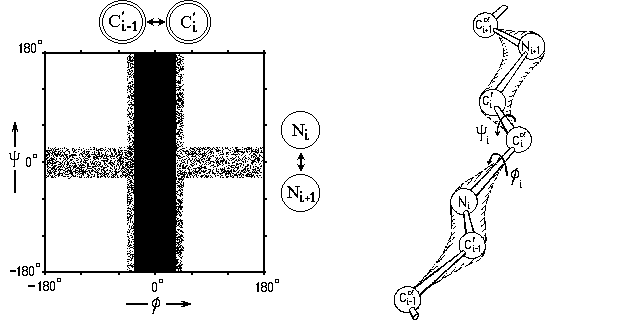
Рис.3-2. Так выглядела бы карта запрещенных ![]() , напряженных
, напряженных ![]() и полностью разрешенных
и полностью разрешенных ![]() конформаций при вращении по углам f, y во фрагменте CaC'N - Ca - C'NCa, если бы ко всем этим атомам не было бы прикреплено больше никаких других (см. справа).
конформаций при вращении по углам f, y во фрагменте CaC'N - Ca - C'NCa, если бы ко всем этим атомам не было бы прикреплено больше никаких других (см. справа).
Следующая схема показывает, что минимальное расстояние между атомами C'i-1 и C'i (при вращении по f) и между атомами Ni и Ni+1 (при вращении по y) — одно и то же, 2.9![]() : это чуть меньше, чем rMIN=3.0
: это чуть меньше, чем rMIN=3.0![]() для C...C взаимодействия (так что здесь cis конформация запрещена), — и чуть больше, чем rMIN=2.7
для C...C взаимодействия (так что здесь cis конформация запрещена), — и чуть больше, чем rMIN=2.7![]() (хоть и меньше, чем r0=3.1
(хоть и меньше, чем r0=3.1![]() ) для N...N взаимодействия (так что здесь cis конформация не запрещена, но напряжена).
) для N...N взаимодействия (так что здесь cis конформация не запрещена, но напряжена).


|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 |
