
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Столь же обычным местом активного центра является место стыка доменов. Рисунок 20-8 показывает активный центр сериновых протеаз типа трипсина, — он находится на стыке двух b-структурных доменов.

Рис.20-8. Положение активного центра в сериновых протеазах типа трипсина. Показаны части активного центра: каталитического центра, где выделены боковые группы "триады переноса заряда" — Ser195 (оранжевый), His57 (синий) и Asp102 (малиновый), и субстрат-связывающего центра, где зеленым изображены NH-группы, образующие оксианионовую дыру, голубым — неспецифическая субстрат-связывающая площадка, и желтым - группы, выстилающие специфический субстрат-связывающий карман.
Здесь естественно начать рассказ о белках, чья главная функция — химически ТРАНСФОРМИРОВАТЬ связавшиеся с ними молекулы.
Сериновые протеазы — классический лекционный объект, используемый для рассказа о простой ферментативной реакции, и я не буду отступать от этой традиции.
Сериновые протеазы разрезают полипептидные цепи, т. е. проводят реакцию

Реакция гидролиза пептидной цепи идет, когда в среде достаточно много воды, и сама по себе, но очень медленно, за многие годы, — то есть гидролиз (при наличии свободной воды) термодинамически выгоден, но требует преодоления очень высокого активационного барьера. Если же свободной воды в среде нет — реакция идет в другую сторону, в сторону синтеза полипептида и выделения воды, но тоже очень медленно.
В присутствии же фермента реакция гидролиза пептида (или, при отсутствии свободной воды, реакции его синтеза из более мелких фрагментов) занимает доли секунды, — то есть фермент резко снижает ее активационный барьер. Посмотрим, как он это делает.
Прежде всего рассмотрим основные компоненты активного центра фермента.
Он состоит из каталитического центра, ответственного за проведение химической трансформации, и субстрат-связывающего центра, призванного правильно подставить субстрат под удар каталитического резака (или, точнее, сварочно/режущего аппарата — так как фермент равно ускоряет и прямую, и обратную реакцию).
Катализ — в сериновых протеазах — непосредственно осуществляется боковой цепью серина (называемого "Ser195" — по его расположению в цепи химотрипсина — во всех белках семейства трипсина; именно он, трипсин, изображен на Рис.20-8). Напомню химическую формулу боковой группы серина: —СН2-ОН. Однако для того, чтобы серин мог катализировать гидролиз — его (серин) нужно подготовить. Пока кислород находится в форме —ОН группы, он не активен, а активным он становится после утери Н+ и перехода в форму —О-. Этим отрывом атома Н от Ser195 занимаются два остальных члена "триады переноса заряда" — His57 (он-то и принимает оторванный атом Н) и "вспомогательный" Asp102. Мутации обоих этих остатков — не говоря уже о мутации каталитического серина — практически губят каталитическую активность трипсиновых протеаз.
Субстрат-связывающий центр состоит (Рис.20-8, 20-9) из оксианионовой дыры, связывающей кислород расщепляемой пептидной группы, из неспецифической пептид-связывающей площадки (отвечающей — вместе с оксианионовой дырой — за то, что расщепляемая пептидная группа займет правильное положение относительно активированного О-атома боковой группы Ser195), и из специфического субстрат-связывающего кармана, отвечающего за распознавание той аминокислоты, по карбоксилу которой производится расщепление пептида.
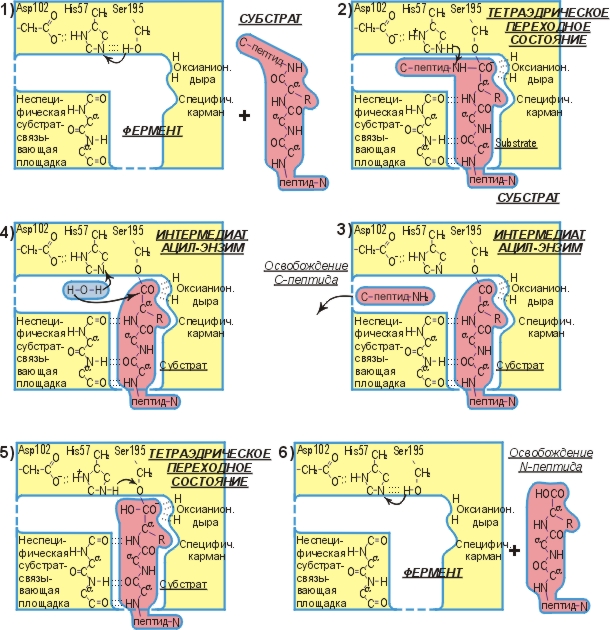
Рис.20-9. Схема ферментативного гидролиза пептида.
Общий ход реакции иллюстрируется рисунком 20-9. Эта схема была получена ценой многолетних исследований многих групп. Для ее построения привлекались и данные по катализу разных субстратов, и по химическим модификациям фермента, а равно и белково-инженерные исследования (которые показали, какие точки фермента вовлечены в катализ), и исследования ферментов в комплексе с нерасщепляемыми аналогами субстратов, и рентгенструктурные работы (показавшие, где образуются водородные связи), и так далее.
Что же делает фермент, как он ускоряет химическую реакцию? Для ответа на этот вопрос давайте сравним ферментативную реакцию (Рис.20-9) с аналогичной реакцией, протекающей спонтанно, — в присутствии воды, но без фермента (Рис.20-10).
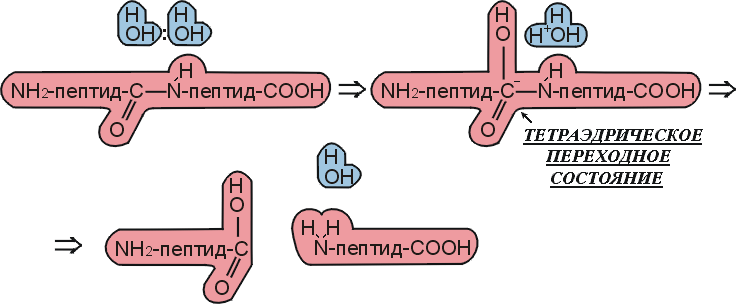
Рис.20-10. Схема спонтанного, не ферментативного гидролиза пептида в воде по тому же, что на Рис.20-9, химическому механизму.
Мы видим, что ферментативная реакция идет в две стадии — сначала отщепляется С-концевой пептид и образуется комплекс N-концевого пептида с О-атомом серина, а затем этот N-пептид заменяет свою связь с О-атомом серина на связь с О-атомом воды. Без фермента же реакция идет в одну стадию, в которой отщепляется С-концевой пептид и образуется комплекс N-концевого пептида с О-атомом воды. Мы видим также, что все эти три реакции идут через тетраэдрический активированный комплекс, — точнее, активированный комплекс, где рядом с тетраэдрическим С-атомом находится донор протона (His+ в ферменте или, в воде, — ОН3+).
То, что на пути ферментативной реакции находится два активированных комплекса (а не один, как в ферментативной) — не может, само по себе, ускорить реакцию. Однако ускоряет ее то, что активированный комплекс в связи с ферментом менее нестабилен (а именно его нестабильность и лимитирует скорость реакции), чем активированный комплекс в воде.
Активированный комплекс на ферменте стабилизируется следующим образом.
Во-первых, отрицательный заряд атома О-, образующийся при тетраэдризации С-атома, на ферменте втягивается в оксианионовую дыру, где стоят два уже нацеленных в эту дыру протона. Связь с этими положительно заряженными протонами понижает свободную энергию этого отрицательного заряда O - и, следовательно, энергию тетраэдризации С-атома — по сравнению с не-ферментативной реакцией, где такой заранее устроенной "дыры" нет, то есть приводит к понижению энергии (энтальпии) переходного состояния, или к так называемому энтальпийному катализу.
Во-вторых, рядом с тетраэдрическим С-атомом стоит, с протоном наготове, His+ — а он примерно столь же стабилен, как "безпротонный" His0. В воде роль донора протона должен играть ион ОН3+, — ион, концентрация которого, вследствие его нестабильности, в воде крайне мала — порядка 10-7 моля на литр. Его "вылавливание" из воды понижает энтропию и соответственно повышает свободную энергию активированного комплекса. Поэтому энтропия препятствует сбору "до кучи" всех компонентов активированного комплекса в воде — и не мешает этому сбору на ферменте, где все эти компоненты уже собраны вместе. При этом энтропия "вылавливания" фермента субстратом оплачивается его прилипанием к субстрат-связывающему карману. Этот облегченный сбор всех компонентов активированного комплекса понижает его свободную энергию на ферменте — по сравнению с оной в не-ферментативной реакции — и приводит к так называемому энтропийному катализу.
В сумме, энтропийная и энтальпийная компоненты катализа ускоряют ферментативную реакцию примерно в 1010 раз по сравнению с не-ферментативной.
Если прилипание субстрата к ферменту достаточно сильно, а концентрация субстрата не слишком низка, то энтропийный катализ понижает наблюдаемый порядок реакции по концентрации субстрата. Теперь ее скорость зависит лишь от концентрации фермента, а от концентрации субстрата (или субстратов) в растворе не зависит. Этот эффект особенно силен тогда, когда речь идет о реакции, в которую вовлечено несколько молекул-субстратов, например — при синтезе пептида из двух фрагментов в отсутствии воды.
Основное и в химическом, и в ферментативном катализе — понижение свободной энергии переходного состояния, — т. е. снижение максимума свободной энергии, преодолеваемого по ходу реакции (Рис.20-11). Снижения этой свободной энергии можно достигнуть за счет энтропии, за счет сбора на ферменте всех необходимых компонентов реакции. И его же можно достигнуть — или усилить — за счет свободной энергии преимущественного связывания именно переходного состояния обрабатываемой молекулы, — а не ее начального состояния — "субстрата" и/или состояния конечного — "продукта". Кстати, если субстрат или продукт будут связываться очень уж сильно (сильнее, чем переходное состояние), — фермент будет ими просто ингибироваться (он потеряет свою функцию "ОТПУСТИТЬ" — и выйдет из игры).
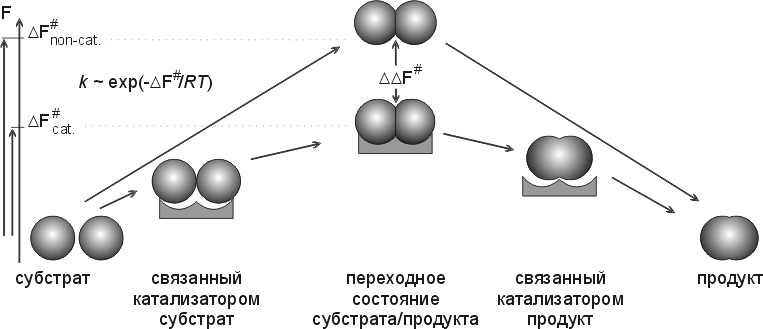
Рис.20-11. Схема, поясняющая важность стабилизации катализатором именно переходного (самого нестабильного по ходу реакции) состояния субстрата/продукта и важность жесткости катализатора для снижения свободно-энергетического барьера реакции (DF#) и увеличения ее скорости (k). Рисунок, для наглядности, изображает просто комплементарность обводов переходного состояния к обводам активного центра фермента, в то время как в действительности основную роль обычно играет комплементарное взаимодействие их электронных облаков. Свободные энергии (F) начального и конечного состояний не меняются катализатором. Рисунок подчеркивает, что ускорение реакции зависит величины DDF#, т. е. от силы связывания ферментом именно переходного состояния: эффективность катализа связана с тем, что оно связывается сильнее, чем и начальный субстрат, и конечный продукт.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 |
