
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Структура N-концевого домена интерферона — одна из первых, более или менее успешно предсказанных до опыта структур белковых молекул.
Хочу подчеркнуть факторы, способствовавшие успеху предсказания: a-спирали предсказаны здесь очень уверенно; и они одинаково расположены в отдаленно-гомологичных цепях, что позволяет им доверять.
Однако столь уверенное и повторяющееся в гомологах предсказание вторичной структуры встречается на практике не часто. Более типичны некоторые разночтения, как в С-концевом домене интерферонов (Рис.19-10), для которого — именно вследствие этого, очень типичного небольшого разночтения — однозначного предсказания укладки цепи получить не удалось.
И еще очень важная вещь для успеха предсказания строения интерферона. Когда мы искали наилучшую упаковку предсказанных a-спиралей, мы могли рационально организовать перебор всех возможных упаковок, так как располагали априорной классификацией a-спиральных комплексов (я об этом уже говорил) — она как раз была разработана к тому времени Мурзиным и мной.
Вернемся к предсказанию вторичной структуры.
Оно носит вероятностный характер: точность распознавания a-, b-структуры и нерегулярных петель белка по его аминокислотной последовательности составляет около 65%.
Можно ли улучшить предсказание вторичной структуры? Да. В методе "PHD" Роста и Сандера (очень его рекомендую, он доступен по Интернет) вторичная структура предсказывается не для одной аминокислотной последовательности, а для набора гомологов. В результате такого подхода случайные погрешности как бы сглаживаются и предсказание становится намного более точным (достигая, в среднем, 72-73% вместо 63-65%).
Итак — несмотря на свою ограниченную точность, предсказание вторичной структуры стало уже, по сути, рутинной процедурой исследования первичной структуры белка.
Перейдем теперь к новой, быстро развивающейся области — к работам по предсказанию пространственных укладок белковых цепей.
Проблему предсказания белковой структуры можно представить себе как проблему выбора той структуры, которая наиболее стабильна для данной аминокислотной последовательности. Проблема заключается в том, откуда взять "возможные белковые структуры". Самый простой, прагматичный ответ — взять из Банка Белковых Структур. Там, конечно, нет еще всех возможных структур — но есть надежда, что там есть примерно половина всех существующих в природе мотивов укладки белковых цепей в домены. Эту надежда — ее обосновал Сайрус Чотиа — основана на том, что вновь расшифровываемые белки (точнее — их домены) все чаще и чаще оказываются похожими на уже расшифрованные.
Кстати, сейчас разворачивается большой проект "Structural Genomics", цель которого — расшифровать пространственное строение хотя бы одного представителя каждого семейства белков (а их — порядка 10000). Когда эта программа будет завершена — для этого придется потратить примерно 10 миллиардов $ за 10 лет — все предсказание структур "новых" белков можно будет — мы надеемся на это — делать по гомологии с уже расшифрованными белками. Однако пока что приходится использовать лишь вероятностные методы предсказания; в настоящее время они — точнее, лучшие из них — дают правильный ответ (для белков, не имеющих видимой гомологии с уже расшифрованными) примерно в 30% случаев.
Итак, предсказывая структуру белка, не имеющего видимой гомологии с белками уже расшифрованными, можно попробовать взять, одну за другой, все пространственные структуры из Банка, наложить (возможно, с некоторыми выпетливаниями, см. Рис.19-12) цепь этого белка на каждую из них, и посмотреть, какая из этих пространственных структур даст — для нашей цепи — наибольший энергетический выигрыш. При этом мы должны разрешать цепи то идти по скелету структуры, то выпетливаться или "сокращать" имеющиеся в скелете выпетливания — если это увеличивает энергетический выигрыш.
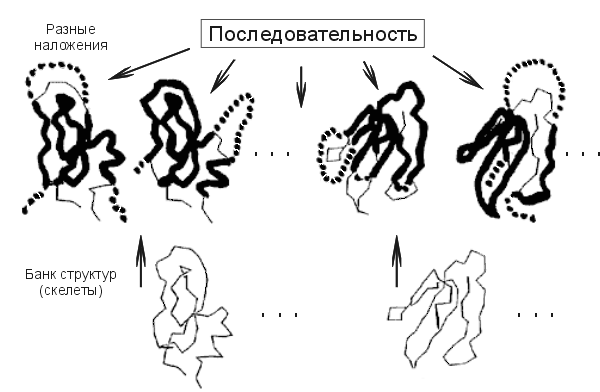
Рис.19-12. Схема, иллюстрирующая идею "протягивания" исследуемой последовательности по Банку Белковых Структур. Жирная линия показывает те участки, где последовательность идет по скелету, пунктир — те, где она выпетливается.
Такой подход называют "методом протягивания" (threading method). Он был предложен нами с в 1990 г. и — независимо, в более простом и более удобном варианте — Д. Айзенбергом и его группой в 1991 г. Сейчас метод протягивания стал весьма популярным методом опознавания структур "новых" белков по их аналогии со "старыми".
В общем, работа по протягиванию напоминает поиск гомологии, — только на этот раз "выравниваются" не две первичные структуры, "новая" и "старая", а "новая" первичная структура со "старым" белковым скелетом.
Прежде, чем перейти к результатам, — хочу подчеркнуть две принципиальные проблемы метода "протягивания". В сущности, аналогичные проблемы, в том или ином виде, возникают в любом "предсказательном" методе.
Во первых, конформацию даже тех кусков цепи, что наложены на скелет, мы знаем с большой погрешностью: ведь мы не знаем конформации боковых групп, — а именно они, в основном, и взаимодействуют. Далее, мы не знаем конформации всех выпетливаний. Оценка показывает, что при протягивании мы знаем примерно половину взаимодействий в белковой цепи, а вторую — не знаем. Значит, опять мы вынуждены судить о структуре белка по части взаимодействий, действующих в его цепи. Значит, опять наши предсказания могут носить только вероятный характер.
Во-вторых, как перебрать все наложения и найти лучшее — или лучшие — среди них? Ведь их, возможных наложений, страшно много... Скажу коротко: мощные математические вычислительные методы для этого уже развиты, но рассказ об этом занял бы слишком много времени. Наверно, имеет все же смысл назвать эти методы по имени — может быть, кому-то из вас пригодится (но большинству, конечно, нет). Итак: динамическое программирование и его вариант — статистическая механика одномерных систем (цепных молекул) — для расчета протягивания цепи через скелет; теория самосогласованного поля — для расчета действующего на цепь молекулярного поля в каждой точке скелета; стохастическая минимизация энергии методом Монте-Карло; а также — разные варианты метода ветвей и границ, и т. д.
Для примера я покажу предсказанную методом протягивания укладку цепи в белке, терминирующем репликацию. Протягивая цепь этого "нового" белка по всем известным трехмерным белковым структурам, Зиппль и его коллеги из Зальцбурга показали, что укладка гистона Н5 наиболее "родственна" этой цепи (Рис.19-13).
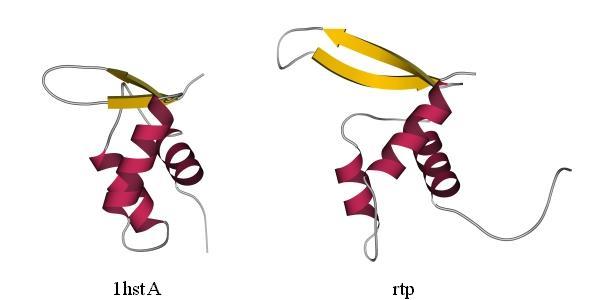
Рис.19-13. Гистон Н5 цыпленка (1hstA, слева) и белок, терминирующий репликацию (rtp, справа); в последнем не показана С-концевая спираль, не имеющая аналога в гистоне Н5. Предсказание сходства укладки цепи в этих двух белках сделано, методом "протягивания", в работе H. Fl![]() ckner, M. Braxenthaler, P. Lackner, M. Jaritz, M. Ortner & M. J.Sippl, Proteins (1995) 23:376-386; это предсказание сделано в ходе "слепого" тестирования предсказательных методов (CASP-1994). Средне-квадратичное отклонение между 65-ю эквивалентными Сa атомами в изображенных структурах — 2.4
ckner, M. Braxenthaler, P. Lackner, M. Jaritz, M. Ortner & M. J.Sippl, Proteins (1995) 23:376-386; это предсказание сделано в ходе "слепого" тестирования предсказательных методов (CASP-1994). Средне-квадратичное отклонение между 65-ю эквивалентными Сa атомами в изображенных структурах — 2.4![]() .
.
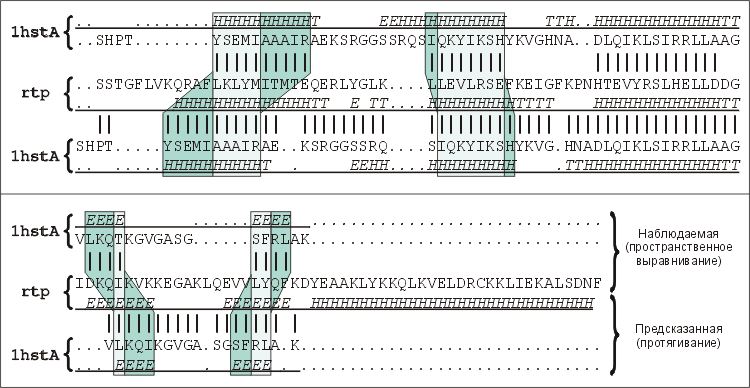
Рис.19-14. Последовательность гистона Н5 (1hstA), выровненная относительно последовательности белка, терминирующего репликацию (rtp) согласно рентгеноструктурным данным ("Наблюдаемая", верхняя пара последовательностей), и она же, выровненная согласно предсказанию методом протягивания ("Предсказанная", нижняя пара). Серыми зонами показаны сдвиги, расхождения этих двух выравниваний. Они хорошо видны по разным сдвигам вторичных структур гистона Н5, приведенных в самой верхней и самой нижней строке в этих двух выравниваниях. Вторичные структуры записаны в подчеркнутых строках (обозначения: Н — a-спираль, Е — b-тяж, Т — изгиб цепи). Точками отмечены делеции, пропуски в первичных и вторичных структурах, внесенные при их выравнивании. Картинка взята из статьи C. M.-R. Lemer, V. J.Rooman & S. J.Wodak, Proteins (1995) 23:337-355, в которой обсуждаются итоги CASP-1994.
Таким образом они правильно опознали мотив укладки цепи белка — терминатора репликации. Правда, предсказанное (при помощи теоретического протягивания) наложение цепи этого белка на структуру гистона Н5 довольно сильно отличается (Рис.19-14) от того, что получается при прямом наложении трехмерных структур этих молекул. Это еще раз показывает, что все погрешности, о которых я говорил по ходу лекций — погрешности в параметрах стабильности, незнание точной конформации петель и т. д. — не позволяют выделить одну уникальную наилучшую структуру, а позволяют в действительности только найти более или менее узкий набор лучших структур. Такой набор "лучших" пространственных структур можно выделить довольно надежно; а вот какая из них окажется, согласно расчету, "наилучшей" — это дело случая. Нативная структура находится где-то в наборе "лучших" структур, она приблизительно соответствует предсказанной ("наилучшей"), — но это все, что можно реально сказать.
Я, конечно, привел только "хорошие" примеры — примеры удавшихся предсказаний. Примеров неудавшихся много больше, и это понятно — ведь, предсказывая, надо выбрать один, или пусть несколько вариантов из бездны оцениваемых. Так что попасть рядом с такой бесконечно малой целью даже в трети или в четверти случаев (так сейчас работают наиболее успешные группы), — огромный успех.
Поэтому методы "протягивания" становятся сейчас рабочим инструментом для опознавания трехмерных структур белковых последовательностей. Чрезвычайно важно, что уже сформирован рецепт — делай так, так и так, и в результате ты получишь одну или несколько структур, среди которых, с довольно высокой вероятностью, будет структура, сходная с трехмерной укладкой рассматриваемой последовательности.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 |
