
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
|
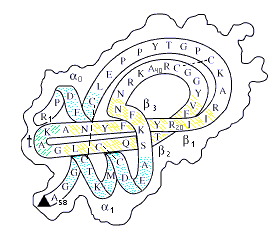
Рис.19-5. Схема первичной и пространственной структуры маленького белка (панкреатического ингибитора трипсина). Ход главной цепи изображен на фоне общего контура молекулы; выделены a-спирали, b-тяжи, резкий поворот цепи (t) и цистеиновые мостики ( - - - ). Так как белок сворачивается сам собой, то — в принципе — все это можно предсказать по одной лишь первичной структуре белка. Боковые группы здесь не показаны, но — в принципе — и их расположение в пространстве тоже можно предсказывать.
Надо сразу сказать, что абсолютно надежных и точных методов предсказания белковых структур сейчас нет.
Причин тому, видимо, две: (а) ограниченная точность энергетических оценок, на которых базируется теоретический расчет белковых структур, и (б) сравнительно малая разность между "правильно" и "неверно" уложенными белковыми цепями. Я хочу особо подчеркнуть последнее обстоятельство, — оно резко отличает ситуацию в белках (и РНК) от ситуации в ДНК, — к большому огорчению людей, занявшихся предсказаниями структур белков по их аминокислотным последовательностям. В ДНК комплементарное спаривание нуклеотидов наблюдается по всей длине двойной спирали, а оно обеспечивается тем, что одна цепь по своей первичной структуре строго комплементарна другой. Это и дает огромный свободно-энергетический выигрыш "правильной" структуры ДНК над всеми "неправильными". А в белках (и РНК) ни какой-либо строгой комплементарности, ни следующего из нее огромного энергетического преимущества "правильной" укладки цепи не наблюдается...
Однако существующие методы дают все более полную и надежную информацию о возможном строении (или, чаще, о возможных вариантах строения) белка или, особенно, его структурных элементов.
В прагматическом аспекте, вопрос о предсказании трехмерного строения белка сейчас ставится следующим образом: похожа ли пространственная структура рассматриваемой белковой последовательности на какую-либо из уже известных пространственных белковых структур? Если да, то как рассматриваемая последовательность вписывается в эту структуру? Если нет, можем ли мы указать какие-либо характерные детали пространственной организации рассматриваемой последовательности?
Я знаю несколько случаев, когда ответы на эти вопросы существенно способствовали экспериментальному определению структуры и/или функции изучаемой аминокислотной последовательности, помогли планировать белково-инженерные эксперименты и так далее. Впрочем, должен сказать, что я бы с большим интересом прочел бы хороший обзор о практическом применении предсказаний белковых структур...
Перейдем теперь к методам предсказания белковых структур. Рассматривая эти методы, я буду уделять особое внимание тем идеям, и в основном физическим идеям, что лежат в их основе.
Есть две стратегии предсказания белковых структур. Согласно первой стратегии, белковая структура ищется как результат кинетического процесса сворачивания.
Согласно второй, — она ищется как структура с минимально возможной для данной цепи свободной энергией.
В принципе, по-видимому, обе эти стратегии могут привести к правильному результату, так как самая стабильная структура белковой цепи, несмотря на опасения Левинталя, образуется достаточно быстро (этому вопросу была посвящена отдельная лекция, и сейчас я позволю себе его не касаться).
Важно, однако, что, рассматривая предсказание белковых структур, мы можем отвлечься от путей сворачивания и рассматривать только результат — стабильную структуру белка. Тем более, что первая стратегия — стратегия имитационного моделирования процесса сворачивания белка — пока существенного прогресса не дала: уж очень сложны все расчеты, и к тому же — все же весьма приблизительны все потенциалы взаимодействий. Вторая стратегия оказалась более успешной.
Начнем с определения стабильной вторичной структуры белка по его аминокислотной последовательности. a-спирали и вытянутые b-участки — важнейшие элементы белковых глобул, во многом определяющие, как вы помните, их общую архитектуру (Рис.19-5).
Забудем пока о том, что белковая цепь свернута в глобулу, т. е. рассмотрим развернутую белковую цепь. Можем ли мы предсказать ее вторичную структуру по аминокислотной последовательности? Оказывается — да, в основном да, пусть и не абсолютно точно.
Прежде всего, — какие аминокислотные остатки будут стабилизировать вторичную структуру — например, a-спираль, — а какие будут разрушать ее?
Эксперименты дают прямой ответ на этот вопрос. Я имею в виду огромную работу по оценке a - и b-образующих способностей аминокислотных остатков, проведенную в группах Шераги, Фасмана, Болдвина, Фершта, Серрано, ДеГрадо, Кима и в ряде других групп (и, в частности, у нас — и ). Кроме того, богатый (и хорошо совпадающий с физико-химическим экспериментом) материал дает статистика аминокислотных последовательностей a - и b-структур в белках. Рисунок 19-6 суммирует важнейшие (те, которые стоит запомнить) из оценок, полученных всеми этими методами.
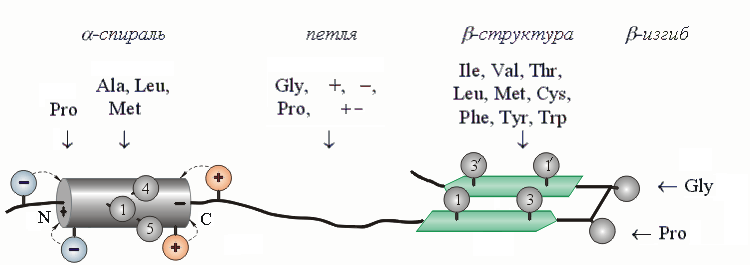
Рис.19-6. Шаблоны a-спирали, петли, b-структуры и b-изгиба — те остатки, которые стабилизируют их или их отдельные части. "+" означает все положительно заряженные аминокислоты, "-" — все отрицательно заряженные, "+ -" — все аминокислоты с диполем в боковой цепи. Показан также стабилизирующий a - и b-структуру порядок чередования гидрофобных групп в цепи (см. нумерованные группы). Такого типа чередование приводит также к образованию гидрофобных и полярных поверхностей a-спиралей и b-тяжей.
Здесь надо, однако, сразу сказать, что все закономерности, наблюдающиеся в структурах белков, носят вероятностный, частотный характер. Несмотря на многочисленные попытки, не удалось выделить никакого четкого "кода" белковых структур — то есть ничего похожего на то четкое A-T и G-C спаривание нуклеотидов, которое свойственно двойной спирали ДНК. Впрочем, надо признать, что уже в РНК — с их разнообразным, в отличие от ДНК, репертуаром пространственных структур — спаривание нуклеотидов происходит далеко не столь однозначно...
Подавляющее большинство из полученных экспериментальных и статистических оценок можно легко понять, исходя из физики и стереохимии аминокислот. Мы уже говорили об этом на одной из прошлых лекций.
Так, Pro не любит входить ни в a-спираль (кроме ее N-концевого витка), ни в b-структуру. Почему? Потому, что у него нет NH-группы, и он не может завязывать соответствующие a - и b-структуре водородные связи; а на N-конце спирали NH-группа таких связей и не должна завязывать, — и Pro там встречается часто, тем более что его угол y уже фиксирован пролиновым кольцом в подходящем положении. По тем же причинам часто встречается Pro и на N-конце изгибов.
А вот Ala стабилизирует a-спираль, — а Gly разрушает и ее, и b-структуру, и способствует образованию клубка. С чем связана эта разница? С тем, что область конформаций, т. е. область допустимых углов f, y для Gly в клубке гораздо больше, чем для Ala, — в то время как допустимые конформации для обоих этих остатков в a-спирали примерно совпадают.
По аналогичным причинам разветвленные остатки — Val, Ile и Thr — больше стабилизируют b-структуру, где их боковые группы имеют 3 разрешенных поворотных изомера, чем a-спираль или клубок, где они имеют только 1 изомер (при каждом значении f и y).
Вообще же гидрофобные группы склонны несколько чаше входить в a - и b-структуру, где они могут слипаться в гидрофобных кластерах (см. Рис.19-6), чем в клубок, где они этого делать не могут. А вот боковые группы с диполями, особенно — в короткой боковой цепи, больше склонны входить в нерегулярные участки цепи, где они могут завязывать дополнительные нерегулярные водородные связи, чем в a - и b-структуры, где доноры и акцепторы водородных связей уже насыщены внутрицепными связями.
Более того, влияние аминокислотных остатков на вторичную структуру можно оценить теоретически. Так, еще до получения экспериментальных оценок, в начале и середине 70-х годов, мы с предсказали, что отрицательно заряженные остатки должны стабилизировать N-конец спирали, притягиваясь к его положительному парциальному заряду, и дестабилизировать ее С-конец, отталкиваясь от отрицательного парциального заряда спирального диполя. Положительно же заряженные остатки должны действовать в прямо противоположном направлении. При этом потенциал каждого такого взаимодействия должен, по теоретической оценке, составлять 1/4 или 1/3 килокалории. Что и было подтверждено экспериментально.
Зная, какие аминокислотные остатки стабилизируют середину спирали, какие — ее N-конец, а какие — C-конец, мы получаем нечто вроде "шаблона" спирали. Шаблон a-спирали можно описать так: если начало фрагмента белковой цепи обогащено отрицательно заряженными группами, да еще там стоит Pro; если середина этого фрагмента обогащена остатками Ala, Leu и Met, а Pro и Gly там нет; и если его С-конец содержит положительно заряженные боковые группы, — то перед нами a-спиральный участок. Кроме того, для стабильности a-спирали полезен, а для ее включения в глобулу — просто необходим определенный (Рис.19-6) порядок чередования гидрофобных групп в цепи: этот порядок способствует слипанию этих групп и, кроме того, приводит к образованию на спирали сплошной гидрофобной поверхности, необходимой для ее прилипания к глобуле.
То есть "шаблон" качественно описывает аминокислотную последовательность, подходящую для образования a-спирали. Чем лучше аминокислотная последовательность удовлетворяет этому шаблону — тем вероятнее спираль в данном месте цепи. Такое же описание — "шаблон" — можно составить и для участков, пригодных для образования b-структуры. А также — для участков, особо пригодных для образования b-изгибов и петель.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 |
