
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Хуже обстоит дело, когда белки в семействе сильно варьируют (Рис.19-2).
В этом случае на помощь приходит компьютер. Разработано множество программ, ищущих гомологии; с ними можно работать по Интернет. Назову только самые популярные из этих программ: BLAST и PSI-BLAST. Все они строят выравнивание (alignment) последовательностей, добиваясь наибольшего сходства между ними. При этом за повышение сходства часто приходится платить "разрывом" последовательностей (см. знаки "-" на Рис.19-2).
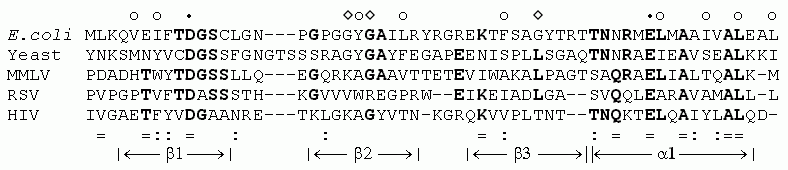
Рис.19-2. Аминокислотные последовательности N-концевых фрагментов рибонуклеаз Н бактерии (E. coli), эукариота (дрожжи, yeast), и трех разных вирусов. Множественное выравнивание делалось так, чтобы не допустить разрывов последовательностей (см. "- - -") внутри a - и b-структурных участков. Жирным шрифтом выделены остатки, идентичные в трех и более из этих пяти последовательностей. Черными точками отмечены остатки активного центра, пустыми кружками и ромбами — остатки, вовлеченные в два гидрофобных ядра этого белка. Внизу отмечены остатки, совпадающие ( = ) и сходные ( : ) у последовательностей из RSV и HIV (помещенных в двух нижних строках выравнивания), а также указана вторичная структура рассматриваемых белков. Картинка, с небольшими изменениями, взята из [7].
Разные программы по-разному оценивают, чего стоит совпадение остатков, чего — сходство, чего — несовпадение, чего — начало разрыва, чего — каждый дополнительный остаток в разрыве. Все эти оценки оптимизируются авторами так, чтобы удовлетворительно выделять белки, сходство которых уже известно из других данных, и потом "зашиваются" в программу. Пользуясь программой, люди ("пользователи") обычно даже не знают, что "хорошо" согласно этой программе, что "плохо", а просто говорят: "установлено, что гомология последовательностей составляет 25%" — имея в виду, что 25% выровненных остатков совпали друг с другом.
Встает вопрос — свидетельствуют ли эти 25% о сходстве последовательностей? Для ответа на этот вопрос необходимо сравнить, пользуясь той же программой, заведомо несходные последовательности. И тут выясняется, что "гомология" несходных белков (Рис.19-3) обычно составляет 10-15%, иногда — 20%, и порой — даже 25%!

Рис.19-3. Выравнивание аминокислотных последовательностей непохожих, негомологичных белков [в данном случае — a-спирального РНК-связывающего белка (rop) и b-структурного белка холодового шока (mjc)] часто дает 10-15% совпадающих аминокислотных остатков [в данном примере — 10 остатков (см. жирный шрифт) из 69, т. е. 14.5%]. Выравнивание сделано программой BLAST.
Эти цифры меняются от программы к программе. Однако накопленный опыт показывает, что тогда, когда "хорошая" (по общему мнению) программа дает совпадение свыше 30-35% остатков, — то выявленной гомологии можно смело доверять (с оговоркой: при длине сравниваемых последовательностей свыше 50, а лучше — 100 остатков). Правда, надо учитывать что 30 — 35% гомологии между последовательностями, верно (как правило) свидетельствуя об их родстве, позволяют правильно наложить друг на друга только 70-80% их пространственных структур, давая неверное предсказание о сходстве остальных 20-30%. А для того, чтобы верно проследить структуру 95% главной цепи "нового" белка, нужно, чтобы его гомология с белком с известной структурой достигала 40 — 50%.
Если же сходство пары последовательностей не превышает 10-15% — то их родство обычно нельзя обнаружить: такое сходство находится на уровне шума (что, однако, не является доказательством, что белки не похожи, не гомологичны — я к этому еще вернусь). А от 15 до 25 и даже до 30% простирается "сумеречная зона": кажется, что белки гомологичны, — но кто поручится?...
К сожалению, все эти цифры не вполне одинаковы у разных программ (и у разных режимов их работы), а к программе они, эти цифры, обычно не прилагаются (они есть в исходных статьях, но кто их читает...), — так что я бы рекомендовал, прежде чем доверяться любой такой программе, проверить ее (именно ее, и именно в используемом Вами режиме) на известных вам белках примерно той же длины (и сходных, и несходных) и понять, "что такое хорошо и что такое плохо" (другой вариант: прочесть исходную статью...).
Больше всего все эти оценки достоверности и недостоверности найденного сходства "плавают" от программы к программе из-за того, что разные авторы по-разному оценивают "штраф" за разрыв последовательности. Если его положить нулевым, то есть позволить делать любые разрывы "бесплатно", — случайно выбранные белковые (и вообще 20-буквенные) последовательности дают сходство на уровне 30-35% (а ДНКовые, 4-буквенные — на уровне 65%)!
Опыт показывает, что оптимальное отделение "похожих" от "непохожих" белковых последовательностей достигается, когда начало разрыва последовательности штрафуется в цену двух или трех дополнительных совпадений аминокислотных остатков, а за удлинение разрыва платится примерно 1/20 — 1/100 этой цены за каждый дополнительный остаток в разрыве.
Я умышленно не говорю ничего о математике, лежащей в основе алгоритмов поиска гомологий. Это нас увело бы слишком далеко. Хочу, однако, произнести ключевые слова: "динамическое программирование". Это — название самого мощного метода, применяемого для оптимизации одномерных систем (а последовательность — система именно одномерная), — в частности, для оптимизации выравнивания одной последовательности относительно другой.
Можно ли распознать гомологичность, родственность последовательностей, если их сходство лежит ниже уровня в 30%, — т. е. в "сумеречной зоне" или даже ниже ее? Можно — но для этого надо сравнивать интересующую нас последовательность со многими последовательностями семейства, и обращать внимание преимущественно на те позиции в цепи, что доказали свою консервативность именно в этом семействе.
Рисунок 19-2 показывает, что рибонуклеаза Н вируса иммунодефицита человека (HIV) имеет не очень высокое — "на уровне шума" — сходство с другими рибонуклеазами Н, если рассматривать всю цепь (так, из 60 выровненных остатков у нее есть всего 9 общих — 15% совпадений — с рибонуклеазой Н из RSV). Однако это сходство проявляется именно в тех ключевых районах, где все остальные рибонуклеазы похожи друг на друга. Это резко повышает достоверность такого сходства. А если еще учесть, что эти "ключевые районы" совпадений охватывают все аминокислотные остатки активного центра, и что сходство концентрируется в участках вторичной структуры, и что оно охватывает около 30% (а не 15%) остатков гидрофобных ядер белка, — высокая достоверность переходит в уверенность в правильном опознании гомологии.
Для опознания гомологии "новых" последовательностей также удобно пользоваться "консенсусными последовательностями" (Рис.19-4), выведенными для уже изученных белковых семейств и подчеркивающими их наиболее консервативные черты. Иногда такие консенсусные последовательности (снабженные данными по частотам встречаемости остатков в каждом месте цепи) называют "профилями первичных структур".
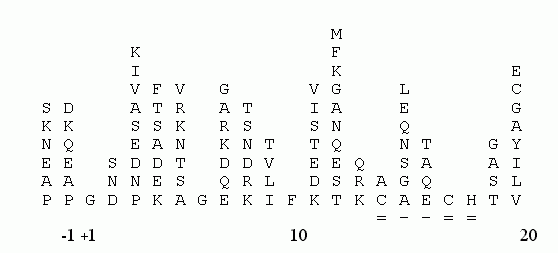
Рис.19-4. Аминокислотный состав различных позиций в N-концевых фрагментах цитохромов c митохондрий эукариот. Самые важные, консервативные остатки цепи определены однозначно. Подчеркнута последовательность "сайта" Cys-X-X-Cys-His, отвечающего за связывание гема в подавляющем большинстве цитохромов (и не только c, и не только эукариот). Выравнивание аминокислотных последовательностей взято из [6].
При опознавании функционального сходства белков следует также обращать внимание на уже установленные для многих функций "сайты" — более или менее короткие последовательности, обеспечивающие эти функции (см. на Рис.19-4 сайт Cys-X-X-Cys-His, связывающий гем в цитохромах). Такие сайты собраны в библиотеки, и их поиском занимаются специальные программы, из которых PROSITE является наиболее популярной.
При установлении структуры "нового" белка по его гомологии с уже изученным надо ясно отдавать себе отчет, что сходство пространственных структур может не распространяться на районы, где последовательности сильно разошлись. В основном это (см. Рис.19-2) районы петель, нерегулярных конформаций белковой цепи. Здесь, с весьма переменным пока успехом, приходится прибегать к конформационным расчетам и другим методам гомологического моделирования, на которых я останавливаться не буду.
Перейдем к методам предсказания пространственной структуры "новых" последовательностей, не имеющих видимой гомологии с уже расшифрованными белками.
К ним относится около 2/3 "новых" последовательностей. Поэтому о пространственной укладке большинства последовательностей, получаемых в ходе выполнения генетических проектов, мы не можем догадаться по их гомологии с белками уже известными — она или слишком слаба для обнаружения, или отсутствует. Тут-то и возникает настоятельная потребность в решении задачи предсказания пространственной структуры — а, в перспективе, и функции белка, — по его аминокислотной последовательности (Рис.19-5).

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 |
